
Une étude cristallographique: superspace description of a commensurate composite cocrystal of 4,4′-dinitrobiphenyl and biphenyl†
Toms
Rekis
 *a,
Sitaram
Ramakrishnan
a,
Surya Rohith
Kotla
a,
Jin-Ke
Bao
a,
Claudio
Eisele
a,
Andreas
Schönleber
a,
Leila
Noohinejad
b,
Carsten
Paulmann
bc,
Martin
Tolkiehn
b and
Sander
van Smaalen
*a
*a,
Sitaram
Ramakrishnan
a,
Surya Rohith
Kotla
a,
Jin-Ke
Bao
a,
Claudio
Eisele
a,
Andreas
Schönleber
a,
Leila
Noohinejad
b,
Carsten
Paulmann
bc,
Martin
Tolkiehn
b and
Sander
van Smaalen
*a
aLaboratory of Crystallography, University of Bayreuth, 95440 Bayreuth, Germany. E-mail: toms.rekis@uni-bayreuth.de; smash@uni-bayreuth.de
bP24, PETRA III, Deutsches Elektronen-Synchrotron, 22607 Hamburg, Germany
cMineralogisch-Petrographisches Institut, Universität Hamburg, 20146 Hamburg, Germany
First published on 26th November 2021
Abstract
We report the structure of a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 cocrystal of 4,4′-dinitrobiphenyl and biphenyl. The cocrystal can be regarded as a commensurate column composite crystal and therefore is described in superspace. The structure is monoclinic at 100 K (superspace group C2(σ10σ3)0), but undergoes a single-crystal-to-single-crystal phase transition upon cooling, which results in a triclinic distortion at 20 K (C1(σ1σ2σ3)0). Unlike similar symmetry-lowering phase transitions, this transition is not found to induce twinning. Other structurally similar cocrystals with 4,4′-dinitrobiphenyl and small organic molecules have been reported in the literature demonstrating that 4,4′-dinitrobiphenyl is prone to form composite crystals.
:1 cocrystal of 4,4′-dinitrobiphenyl and biphenyl. The cocrystal can be regarded as a commensurate column composite crystal and therefore is described in superspace. The structure is monoclinic at 100 K (superspace group C2(σ10σ3)0), but undergoes a single-crystal-to-single-crystal phase transition upon cooling, which results in a triclinic distortion at 20 K (C1(σ1σ2σ3)0). Unlike similar symmetry-lowering phase transitions, this transition is not found to induce twinning. Other structurally similar cocrystals with 4,4′-dinitrobiphenyl and small organic molecules have been reported in the literature demonstrating that 4,4′-dinitrobiphenyl is prone to form composite crystals.
1 Introduction
An incommensurate or a commensurate composite crystal is a single thermodynamic phase containing at least two components, each of which is arranged in space according to its own lattice.1–4 Sometimes also the term intergrowth compound is used to refer to composite crystals.5,6For two or more different lattices to define a single crystalline phase there are some space-fitting requirements. They follow from the requirement that the unit cell of one component (hereafter denoted as subsystem) must be empty at places where atoms of the other subsystems exist (compare Fig. 1(1) and (2)). Accordingly, two types of composite crystals can be distinguished:4 1) layer composites, and 2) column composites. For the former ones two (or more) subsystems form an interleaved layer structure (see Fig. 1 for schematic examples). The space-fitting requirement for such structures is that c* vectors of both of the subsystems must be identical (if c* is the stacking direction of the layers).
 | ||
| Fig. 1 Schematic representation of incommensurate layer composites. (1–3): The unit cell of one subsystem contains a layer motif (atomic layers parallel to a, b with aA ≠ aB and aA ≠ aC, but with aA, aB, aC and b in a single plane) and empty space to accommodate a layer from another subsystem. (4 and 5): To fulfill the space-fitting requirement for a layer composite structure it is necessary that both c* vectors are identical (notice that c* is perpendicular to a and to b). | ||
The other type of composite crystals — column composites are interleaved columnar structures (see Fig. 2). For such structures the space-fitting requirements are that the vectors  and
and  are identical as well as the vectors
are identical as well as the vectors  and
and 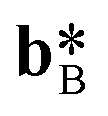 and are identical (if the columns are parallel to c). The columns of each subsystem are spatially distributed on a common grid when projected on the a*b* plane.
and are identical (if the columns are parallel to c). The columns of each subsystem are spatially distributed on a common grid when projected on the a*b* plane.
 | ||
| Fig. 2 Schematic representation of a commensurate (AB) and an incommensurate (AC) column composite. Columns are arranged on a common grid on (a, b) to fulfill the space-fitting requirement. The periodicities of both columns along the c axis are generally not commensurate (see AC), but in case they are — a supercell can be chosen to describe the structure in 3-dimensional space (see AB). | ||
The very essence of composite crystals is that, apart from the space-fitting requirements the lattices of the respective subsystems are rationally independent with each other. Composite crystals are usually incommensurate and therefore non-stoichiometric (regarding their constituent components). For incommensurate layer composites the term misfit layer compound is frequently used.3,7–9
The structures of incommensurate composite crystals can only be described using the superspace formalism.1,2,4 It can happen, however, that supercell lattices can be derived from the respective subsystem lattices so that they match perfectly10 (see Fig. 2(middle) for an example in which cA:cB is a rational number 3:4). In that case the composite crystal is commensurate and a conventional 3-dimensional approach can be used to describe its structure with respect to the supercell.11 Nevertheless, the superspace formalism is often beneficial for understanding commensurate composites and commensurately modulated structures in general.12–15 For example, transitions to structurally similar but incommensurate phases might be induced by varying temperature and pressure.10 Consistency in structure descriptions is crucial for understanding solids.
In this study, a commensurate composite 3:1 cocrystal of 4,4′-dinitrobiphenyl and biphenyl (Fig. 3) has been described in superspace.
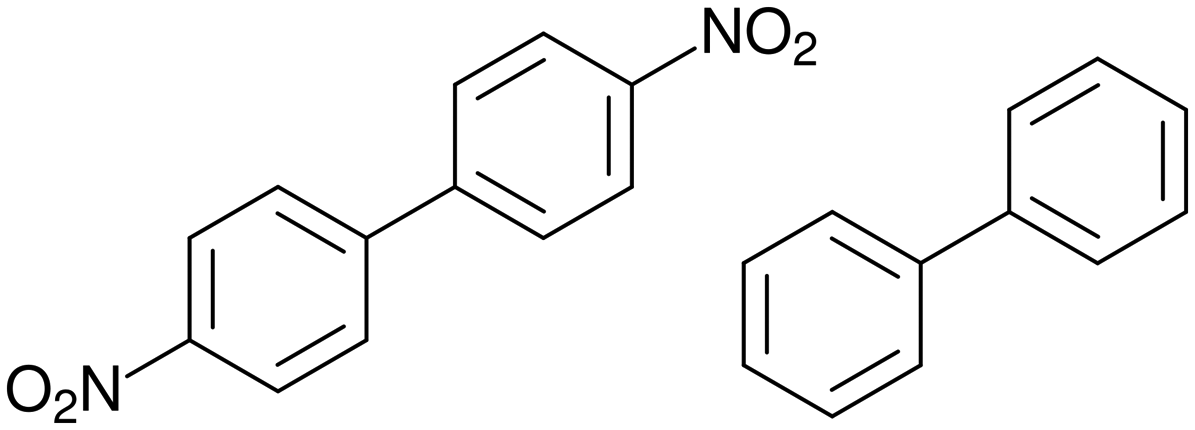 | ||
| Fig. 3 Molecular formulas of 4,4′-dinitrobiphenyl and biphenyl. | ||
The structure of this two-component compound has been studied in 1948.16 However, no correct space group nor the atomic positions were derived. Furthermore, similar cocrystals (between 4,4′-dinitrobiphenyl and other biphenyl derivatives) have been studied at that time some of which apparently being incommensurate.17–19 We show that the structure of the 4,4′-dinitrobiphenyl and biphenyl cocrystal is monoclinic at 100 K and that there is a single-crystal-to-single-crystal phase transition at a lower temperature resulting in a triclinically distorted structure at 20 K.
2 Experimental section
2.1 Single crystals
4,4′-Dinitrobiphenyl (99%) and biphenyl (99.5%) were purchased from Enamine Ltd. and Sigma-Aldrich, respectively. Single crystals were obtained by slow evaporation from an acetone solution.2.2 X-ray diffraction data collection
The diffraction data were collected at beamline P24 of PETRA-III at the German Electron Synchrotron (DESY) facility in Hamburg, Germany. A wavelength of 0.5000 Å was used. The data were recorded on a Pilatus 1M CdTe detector. A total of 10 runs (ϕ-scans) were measured. For each run 3620 frames were collected with an exposure time of 0.1 s per 0.1 deg. The runs were binned to a single run and then each consecutive 10 frames were again binned to obtain 362 frames of 10 s exposure per 1 deg rotation. The crystal was measured at 100 and 20 K.2.3 Data integration
CrysAlisPro 39.46 software20 was used for data integration. Empirical absorption correction using spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm, was applied.2.4 Structure refinement
The structure at 100 K was solved for the supercell in C2, employing SUPERFLIP software.21 Structure refinements were performed with JANA2006.22 Based on the 3D model, a refinement in (3 + 1)D was set up and refined. For hydrogen atoms the riding model was used in the refinements.The (3 + 1)D model was then used for the data measured at 20 K. The R1 converged to a value of 0.0965, which seemed to be unsatisfactory. Triclinic distortion of the monoclinic lattice with possible twinning was considered. The data were averaged according to the Laue class ![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , the contents of the asymmetric unit were doubled using a 2-fold rotation symmetry along the former monoclinic axis and then refined as triclinic. The R1 value decreased to 0.0479.
, the contents of the asymmetric unit were doubled using a 2-fold rotation symmetry along the former monoclinic axis and then refined as triclinic. The R1 value decreased to 0.0479.
3 Results and discussion
3.1 Structures of the cocrystal phases
4,4′-Dinitrobiphenyl and biphenyl forms a 3:1 cocrystal that is a commensurate composite crystal. At 100 K, the unit cells of both subsystems are monoclinic and the superspace group of the composite crystal is C2(σ10σ3)0 (see Table 1 for structural and refinement data). Subsystem A contains half a 4,4′-dinitrobiphenyl molecule in the asymmetric unit and subsystem B contains half a biphenyl molecule in the asymmetric unit. The 4,4′-dinitrobiphenyl molecules are stacked face-to-face forming infinite columns, while the biphenyl molecules form infinite chains with the long molecular axis approximately parallel to the chain direction, i.e. c-axis (see Fig. 4).
:biphenyl 3:1 cocrystal phases (obs. reflections: [F2 > 3σ(F2)])
| Crystal data | ||
|---|---|---|
| T/K | 100 | 20 |
| Formula | 3(C6H8N2O4)·C6H10 | |
| Crystal system | Monoclinic | Triclinic |
| Superspace group | C2(σ10σ3)0 | C1(σ1σ2σ3)0 |
| q | (0, 0, 1/3) | (0, 0, 1/3) |
| t 0 | 0 | 0 |
| Subsystem A | ||
|---|---|---|
| a/Å | 19.7947(4) | 19.7858(8) |
| b/Å | 9.4219(2) | 9.3970(5) |
| c/Å | 3.5826(1) | 3.5684(2) |
| α/° | 90 | 89.986(4) |
| β/° | 98.7506(16) | 98.729(4) |
| γ/° | 90 | 89.982(4) |
| V A/Å3 | 660.39(3) | 655.78(6) |
Z
A, 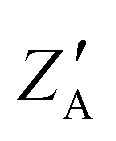 |
2, 0.5 | 2, 1 |
| Subsystem B | ||
|---|---|---|
| W |
|
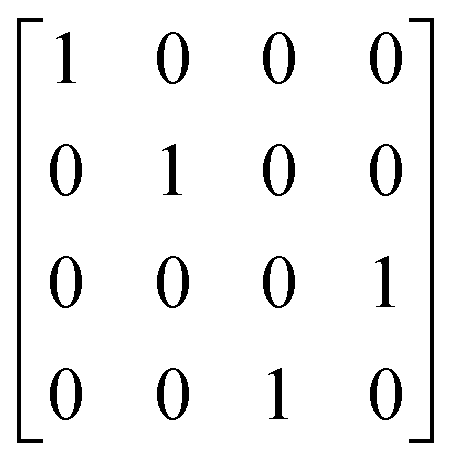
|
| V B/Å3 | 3VA | 3VA |
Z
B, 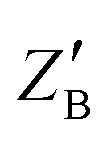 |
2, 0.5 | 2, 1 |
| F(000) | 307 | 307 |
| D x /g cm−3 | 0.8709 | 0.8770 |
| μ/mm | 0.031 | 0.032 |
| Data processing | ||
|---|---|---|
| Laue class | 2/m | |
| R int (obs.) | 0.0661 | 0.0547 |
| No. of refl. | 19670 |
16663 |
| No. of unique refl. | 4533 | 8018 |
| No. of obs. refl. | 4021 | 7100 |
| Resolution limit/Å−1 | 0.65 | 0.65 |
| Refinement | ||
|---|---|---|
| Refinement method | Full-matrix least-squares on F | |
| R 1 (obs.) | 0.0539 | 0.0479 |
| R m=0 (obs.) | 0.0551 | 0.0484 |
| R m=±1 (obs.) | 0.0527 | 0.0475 |
| wR (all) | 0.0734 | 0.0625 |
| No. of parameters | 243 | 483 |
| H-Atom treatment | Constr. | Constr. |
| Weighting scheme | w = 1/(σ2(F) + 0.0001F2) | |
| Δρmax/e Å−3 | 0.32 | 0.45 |
| Δρmin/e Å−3 | −0.20 | −0.34 |
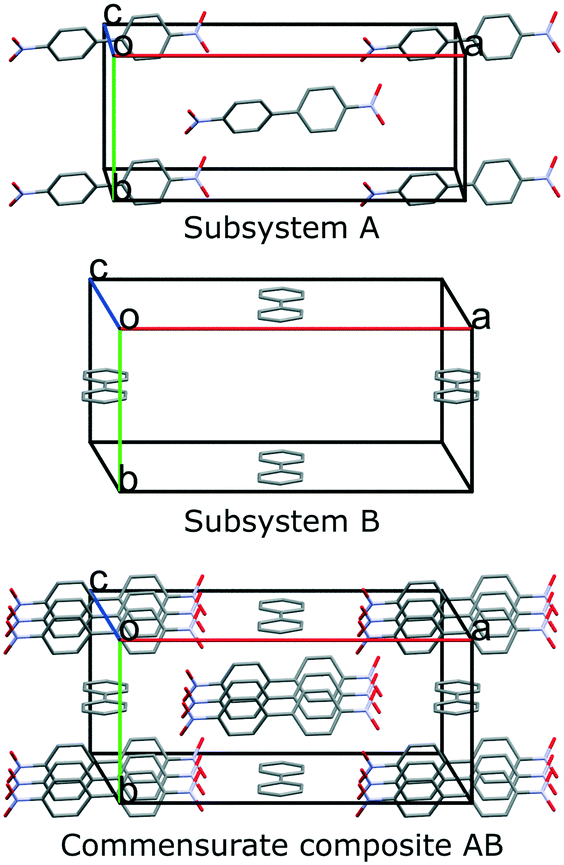 | ||
| Fig. 4 Unit cells of both subsystems and the supercell of the commensurate composite cocrystal. | ||
For column composites one-dimensional modulations can occur in each of the sublattices due to interlattice interaction perturbations.2 For the studied 4,4′-dinitrobiphenyl:biphenyl cocrystal a commensurate modulation in 4,4′-dinitrobiphenyl stacks is present (see Fig. 5). There is a significant displacement of the molecule along the a-axis. This can be summarized in a t-plot depicted in Fig. 6. For the rest of the 4,4′-dinitrobiphenyl atoms the corresponding t-plots are very similar as there is no major variation in molecular conformation. The conformation of the 4,4′-dinitrobiphenyl molecule can be described with two torsion angles — one between the two phenyl rings, and the other one between the NO2 group and the phenyl ring (even though there are two NO2 groups, the molecule has a 2-fold rotation symmetry coinciding with the respective symmetry operator of the (super)space group, i.e. Z′ = 0.5). These torsion angle values are listed in Table 2. Regarding the biphenyl molecule, there is a small deviation from planarity with the torsion angle between the phenyl rings being 1.3(5)°.
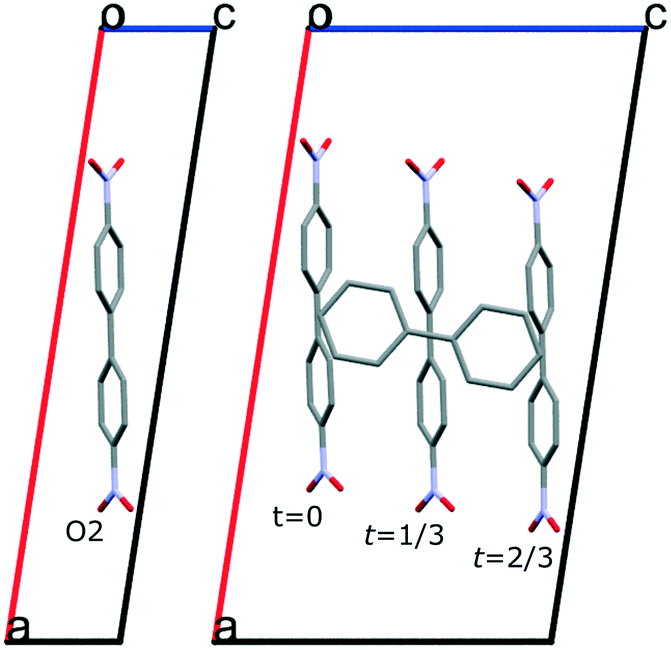 | ||
| Fig. 5 Basic cell of subsystem A (4,4′-dinitrobiphenyl) and the supercell of the commensurate composite cocrystal showing a positional modulation of the 4,4′-dinitrobiphenyl molecules caused by interlattice interaction perturbations. | ||
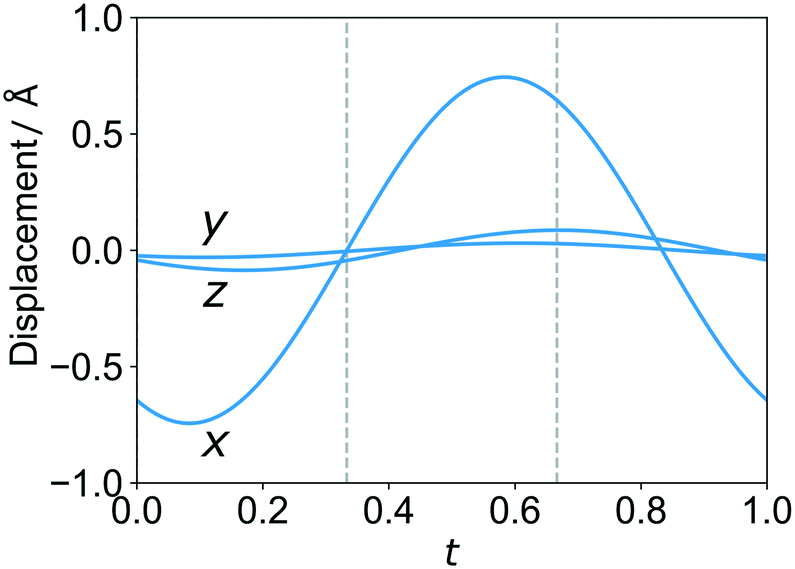 | ||
| Fig. 6 t-Plot of atom O2 displacement (commensurate t sections indicated with dashed lines). The largest displacement from the basic position is present for coordinate x, while the modulation of coordinates y and z is small. | ||
| Torsion/° | Monoclinic | Triclinic | ||||
|---|---|---|---|---|---|---|
| t = 0 | t = 1/3 | t = 2/3 | t = 0 | t = 1/3 | t = 2/3 | |
| Ph–Ph | −35.0(3) | −36.0(3) | −35.0(3) | −32.6(3) | −36.2(3) | −36.4(3) |
| Ph–NO2 | −10.8(3) | −11.4(3) | −3.5(3) | −10.9(4) | −12.2(4) | −3.5(4) |
Ph–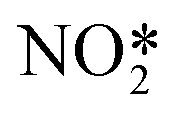 |
−10.8(3) | −11.4(3) | −3.5(3) | −11.0(4) | −12.0(4) | −3.6(4) |
The superspace approach allows to use fewer refinement parameters. In the supercell, there are 33 non-hydrogen atoms present and each of them requires 3 positional and 6 anisotropic displacement parameters (ADPs) which totals 297 refinable parameters. In the (3 + 1)-dimensional structure description, the atomic positions and the anisotropic displacement are described using a set of basic parameters and parametrized modulation functions. The maximum number of parameters, which does not cause singularity during the refinement, is the same for such a commensurate case. However, e.g. the anisotropic displacement modulation functions can be fitted with fewer parameters still resulting in a good description of the electron density. Moreover, such a description is still physically meaningful and sensible. By using a smaller number of ADPs, it was possible to reduce the number of parameters to 243 from the 297‡ required to describe the supercell in 3-dimensional space. Using all the allowed parameters to describe the anisotropic displacement surely reduces the R1 value, however, this reduction is insignificant (from 0.0539 to 0.0516). Another benefit of the superspace approach is that for each parameter and average value, referring to the basic structure, is refined in conjunction with modulation parameters. The latter are usually small thus making the refinement more stable.
As mentioned in the Introduction section, the room temperature (supercell) structure of this cocrystal has been studied in the early days of crystallography.16 No atomic positions were provided and the space group was concluded to be Cm. Our present structural study shows that there are no mirror planes present, i.e. the true space group of the supercell is C2. A structure with mirror symmetry elements would be spatially possible, but in that case all the molecules were completely planar (with the discussed torsion angles of the biphenyl and 4,4′-dinitrobiphenyl molecules being 0°).
Furthermore, we discovered that there is a single-crystal-to-single-crystal phase transition occurring upon cooling for this material. At 20 K the structure is a triclinic distortion of the above described monoclinic phase (superspace group C1(σ1σ2σ3)0, see Table 1). The distortion affects the 4,4′-dinitrobiphenyl molecules for which there is no longer 2-fold rotational symmetry imposed by the (super)space group. The corresponding phenyl–NO2 torsion angles are slightly different in the triclinic phase (see Table 2). Furthermore, the triclinic symmetry is corroborated by decrease in R1 value from 0.0965 to 0.0479 for the monoclinic and triclinic refinements of the 20 K data, respectively. Moreover, α and γ angles are significantly distorted from the monoclinic symmetry (see Table 1).
Lowering of symmetry at low temperatures without major reorganization of the atomic arrangement has long been known. Usually it is accompanied with pseudomerohedral twinning. The twin volume fractions are often equal due to the probabilistic nature of the mechanism behind such a phase transition. Some mechanical factors may cause deviations from the 0.5:0.5 twin volume ratio. In this case, the refinement of the twin volume fraction of the second domain (twvol2; with twvol1 = 1 − twvol2) soon reached a value close to 0 and was unstable. It indicates that by chance only one of the two possible twin domains occurred or its volume fraction is so close to unity that it is impossible to quantify the ratio reliably.
3.2 Other similar cocrystals
4,4′-Dinitrobiphenyl is prone to form structurally similar cocrystals with several substituted biphenyls and urea. The cocrystal with urea (refcode in CSD: BAFWIO) has a stoichiometry of 1:1, it crystallizes in P212121 and is referred to as clathrate.23 The organization of the structure is nevertheless identical to the 3:1 cocrystal of 4,4′-dinitrobiphenyl and biphenyl.
Furthermore, there is a 3:1 cocrystal of 4,4′-dinitrobiphenyl and 4-hydroxybiphenyl (CIMCIK), for which the structure is even more similar to the presently studied material.17,24 In the original study from 1946, monoclinic symmetry was considered and the structure was reportedly solved in space group Cm.17 Around 40 years later this structure was redetermined to show that there is no mirror plane present as the 4,4′-dinitrobiphenyl molecules are twisted and not planar. Furthermore, the redetermined structure is reported as triclinic and treated in a primitive unit cell (space group P1).24 The corresponding C-centered unit cell is nearly monoclinic with α and γ angles of 90.15(2)° and 90.20(2)°, respectively. This is consistent with the observed triclinic distortion of the 4,4′-dinitrobiphenyl – biphenyl cocrystal at low temperature presented in this study. The best overlay of the asymmetric units of the triclinic supercells are depicted in Fig. 7.
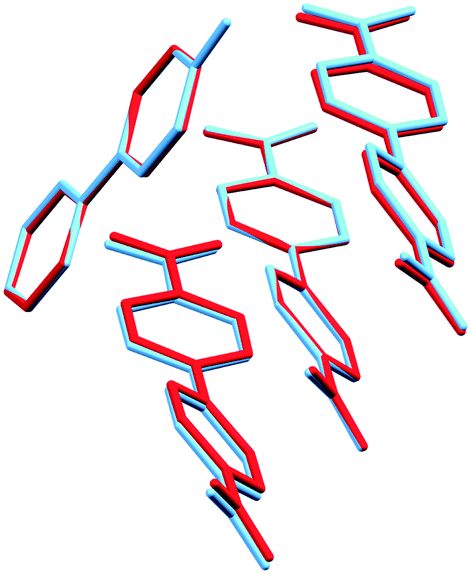 | ||
| Fig. 7 Best overlay of the asymmetric units of the 4,4′-dinitrobiphenyl – biphenyl (red) and 4,4′-dinitrobiphenyl – 4-hydroxybiphenyl (blue) cocrystal triclinic supercells. | ||
In addition, there are 5 other cocrystals with biphenyl derivatives reported having identical structural organization that can be regarded as composite crystals. Those are 4,4′-dinitrobiphenyl cocrystals with benzidine, N,N,N′,N′-tetramethylbenzidine, 4,4′-dihydroxybiphenyl, 4-iodobiphenyl, and 4-bromobiphenyl.18 The stoichiometries vary according to the guest component molecule length (1:3, 1:4, 2:7). Cocrystals with 4-iodobiphenyl and 4-bromobiphenyl apparently feature incommensurate modulations and could be one of the first organic aperiodic crystals ever reported.15 Structural modulation was, of course, not recognized in the original work due to the concept of aperiodic crystallography not being formulated by the time. The diffraction patterns with satellite reflections were referred to demonstrating ‘periodic errors in the crystal lattice’.19
4 Conclusions
4,4′-Dinitrobiphenyl forms a cocrystal with biphenyl that can be regarded as a composite crystal and thus described in superspace. The molecule is prone to form structurally similar composite crystals with several other molecules among which there are substituted biphenyls and urea. The diverse variety of guest molecules that can fill the isostructural channel network formed by 4,4′-dinitrobiphenyl confirms the composite crystal nature of these materials. This knowledge could be utilized in crystal engineering, for example, for designing polar materials for nonlinear optical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Diffraction experiments with synchrotron radiation have been performed at beamline P24 of PETRA-III at DESY in Hamburg, Germany. Funding for this research was provided by the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) 389490692.Notes and references
- P. Coppens, K. Maly and V. Petříček, Mol. Cryst. Liq. Cryst., 1990, 181, 81–90 CrossRef CAS
.
- V. Petříček, K. Maly, P. Coppens, X. Bu, I. Cisarova and A. Frost-Jensen, Acta Crystallogr., Sect. A: Found. Crystallogr., 1991, 47, 210–216 CrossRef
- A. Meerschaut, Curr. Opin. Solid State Mater. Sci., 1996, 1, 250–259 CrossRef CAS
-
S. van Smaalen, Incommensurate Crystallography, Oxford University Press, 2007 Search PubMed
- H.-J. Lamfers, A. Meetsma, S. van Smaalen, J. L. de Boer and K. Huml, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 15697–15706 CrossRef CAS PubMed
- R. Lefort, J. Etrillard, B. Toudic, F. Guillaume, T. Breczewski and P. Bourges, Phys. Rev. Lett., 1996, 77, 4027–4030 CrossRef CAS PubMed
- J. Rouxel, A. Meerschaut and G. Wiegers, J. Alloys Compd., 1995, 229, 144–157 CrossRef CAS
- R. T. Leriche, A. Palacio-Morales, M. Campetella, C. Tresca, S. Sasaki, C. Brun, F. Debontridder, P. David, I. Arfaoui, O. Šofranko, T. Samuely, G. Kremer, C. Monney, T. Jaouen, L. Cario, M. Calandra and T. Cren, Adv. Funct. Mater., 2021, 31, 2007706 CrossRef CAS
-
Y. Ren, PhD thesis, University of Groningen, 1996 Search PubMed
- A. Arakcheeva, G. Chapuis and V. Grinevitch, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 1–7 CrossRef PubMed
- K. Fichtner, Comput. Math. with Appl., 1986, 12, 751–762 CrossRef
- N. B. Bolotina, Crystallogr. Rep., 2007, 52, 647–658 CrossRef CAS
- A. Schönleber, P. Pattison and G. Chapuis, Z. Kristallogr. Cryst. Mater., 2003, 218, 507–513 CrossRef
- T. Rekis, A. Schönleber, L. Noohinejad, M. Tolkiehn, C. Paulmann and S. van Smaalen, Cryst. Growth Des., 2021, 21, 2324–2331 CrossRef CAS
- C. B. Pinheiro and A. M. Abakumov, IUCrJ, 2015, 2, 137–154 CrossRef CAS PubMed
- J. N. van Niekerk and D. H. Saunder, Acta Crystallogr., 1948, 1, 44 CrossRef CAS
- D. H. Saunder, Proc. R. Soc. Lond., A Math. Phys. Sci., 1946, 188, 31–51 CAS
- D. H. Saunder, Proc. R. Soc. Lond., A Math. Phys. Sci., 1947, 190, 508–517 CAS
- R. W. James and D. H. Saunder, Proc. R. Soc. Lond., A Math. Phys. Sci., 1947, 190, 518–533 CAS
- R. O. Diffraction, CrysAlis PRO, Rigaku Oxford Diffraction, 2018 Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS
- V. Petříček, M. Dušek and L. Palatinus, Z. Kristallogr. Cryst. Mater., 2014, 229, 345–352 CrossRef
- R. Thaimattam, D. Shekhar Reddy, F. Xue, T. C. W. Mak, A. Nangia and G. R. Desiraju, J. Chem. Soc., Perkin Trans. 2, 1998, 1783–1790 RSC
- C. P. Brock and K. L. Haller, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 1387–1390 CrossRef
Footnotes |
| † CCDC 2108343 and 2108344. For crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce01223a |
| ‡ In both cases an additional parameter is needed to refine the scale, but the parameter y for one atom needs to be fixed to fix the origin for the (super)space group C2 or C2(σ10σ3)0. |
| This journal is © The Royal Society of Chemistry 2022 |