DOI:
10.1039/D1CE01425K
(Paper)
CrystEngComm, 2022,
24, 504-511
Crystallization kinetics of amorphous red phosphorus to black phosphorus by chemical vapor transport†
Received
22nd October 2021
, Accepted 23rd November 2021
First published on 23rd November 2021
Abstract
Black phosphorus (BP) is an emerging two-dimensional semiconductor material, which has interesting structural features and broad application prospects in the fields of optoelectronic devices, energy storage, and biomedicine. However, a large amount of evidence is still needed to clarify the potential mechanism for the synthesis of high-quality BP crystals in chemical vapor transport (CVT) reactions. In this work, high-purity BP crystals were synthesized by the CVT method in an amorphous red phosphorus (aRP), Sn and I2 system. We found that the synthesis of BP experienced the three stage structural transformation of aRP–P4-Hittorf's phosphorus (HP)–BP. First, the phase transition of aRP and P4 molecules was reversible in the sublimation stage; then, the P4 molecules were gradually transformed into the HP structure in the transportation and deposition stage; however, the nucleation of BP quickly inhibited the growth of HP and completed the phase transition quickly; finally, the remaining P4 molecules were transformed into the BP structure, which promoted the growth of BP crystals. Crystallization kinetics explained the nucleation mechanism during the formation of BP, and the competitive nucleation between HP and BP provided the possibility for the bottom-up preparation of BP nanobelts.
1. Introduction
In recent years, phosphorene has attracted widespread attention in the field of two-dimensional materials due to its unique physical and chemical properties.1,2 Particularly, the excellent anisotropy,3–5 high carrier mobility6 and thermoelectric capabilities7 of phosphorene have led to in-depth research by researchers in the fields of lithium-ion batteries,8–10 field-effect transistors,11 and cancer cell therapy.12–14 Stripping off large BP crystals is the main way to obtain high-quality phosphorene.15–17 So far, synthetic methods for preparing BP include high-temperature and high-pressure conversion of white phosphorus (WP),18 bismuth reflux,19 sonochemical20 and chemical vapor transport (CVT).21,22 Among them, the optimized CVT method, in which Sn and I2 are used as mineralizers, is considered to be the easiest way to synthesize high-purity BP crystals.23,24
Exploring the synthesis mechanism of BP has guiding significance for the controllable synthesis of micro-nano structures. Since the aRP–Sn–I system was first developed for the preparation of BP crystals by Wang's and Yan's group, numerous efforts have been made to explore the growth mechanism of BP crystals during the CVT reaction.25,26 Basically, the CVT reaction is divided into three processes: (1) sublimation, (2) transportation, and (3) deposition.23 The whole reaction process thermodynamically follows Le Chatelier's principle.27 However, the growth mechanism of BP has been controversial in the two processes of transportation and deposition. Initially, Hittorf's phosphorus (HP) was considered to be the intermediate phase of amorphous red phosphorus (aRP) to BP, in which aRP was first transformed into the HP phase, and then from HP to the BP phase.29,30 Unfortunately, this mechanism was gradually being ignored due to the low purity of the growth product.23 Subsequently, researchers found that the ternary compounds Sn24P19.3I8 and BP had topological features near nucleation, and speculated that the inherent P vacancies of Sn24P19.3I8 promoted the growth of BP crystals.21,28,29 However, in recent reports, the appearance of crystalline red phosphorus along with the growth of BP once again overshadows the growth mechanism of BP.24,31
Herein, we report the crystallization kinetics of high-quality BP crystals synthesized by the CVT reaction in the aRP–Sn–I system. The nucleating agent structure, nucleation temperature and nucleation gradient of BP were studied to reveal the aRP–P4-HP–BP three-stage phase transition and nucleation mechanism between aRP and BP. The competitive nucleation between HP and BP provides an idea for the bottom-up preparation of BP nanostructures.
2. Experimental section
2.1 Typical preparation methods of BP
The commercial aRP, Sn and I2 were vacuum sealed in a glass ampoule and placed in a muffle furnace for reaction. The ampoule was heated to 620 °C at 6 °C per minute, cooled to 485 °C at 0.3 °C per minute, held for 2 h, and cooled to 120 °C at 1 °C per minute. Finally, the equipment naturally dropped to ambient temperature. The divergent ribbon product was separated into BP crystals and nucleating agent by simple mechanical separation in the glove box.
2.2 Reaction interruption experiments
In order to explore the change of nucleation and growth of BP at different temperatures, a series of interrupt experiments were conducted. After the glass ampoule containing the reactants was heated to 620 °C, the temperature was normally lowered to the specified temperature. In order to avoid the reaction between the intermediate product and the gaseous substance, the high temperature section of the ampoule was rapidly cooled, so that the gas in the ampoule was rapidly condensed in the low temperature area. The intermediate product and gas condensate were separated in a glove box with full of nitrogen. It is worth noting that the condensate was orange-red, but it was easy to burn under an atmosphere with a lot of white smoke. Similar to white phosphorus, the color changed under the influence of light and mineralization.
2.3 Instrument characterization
Powder X-ray diffraction (XRD) patterns were recorded using a PD3200 (40 KV, 30 mA) X-ray diffractometer with Cu Kα radiation (λ = 1.5406 Å). Raman spectra were acquired on a laser microscopic Raman imaging spectrometer (DXRxi) with a 532 nm laser. Scanning electron microscopy (SEM) images and the corresponding energy-dispersive X-ray spectroscopy (EDX) analysis were carried out in a JIB-4700F (JEOL) scanning electron microscope. Low-magnification TEM images were taken using JEM-2100Plus (JEOL Co., Ltd.) with an accelerating voltage of 200 kV. High-resolution TEM measurements were performed on a JEM-ARM 300F (Cold Field Emission Gun) at an acceleration voltage of 200 kV. Photographs were taken with an optical microscope and digital camera.
3. Results and discussion
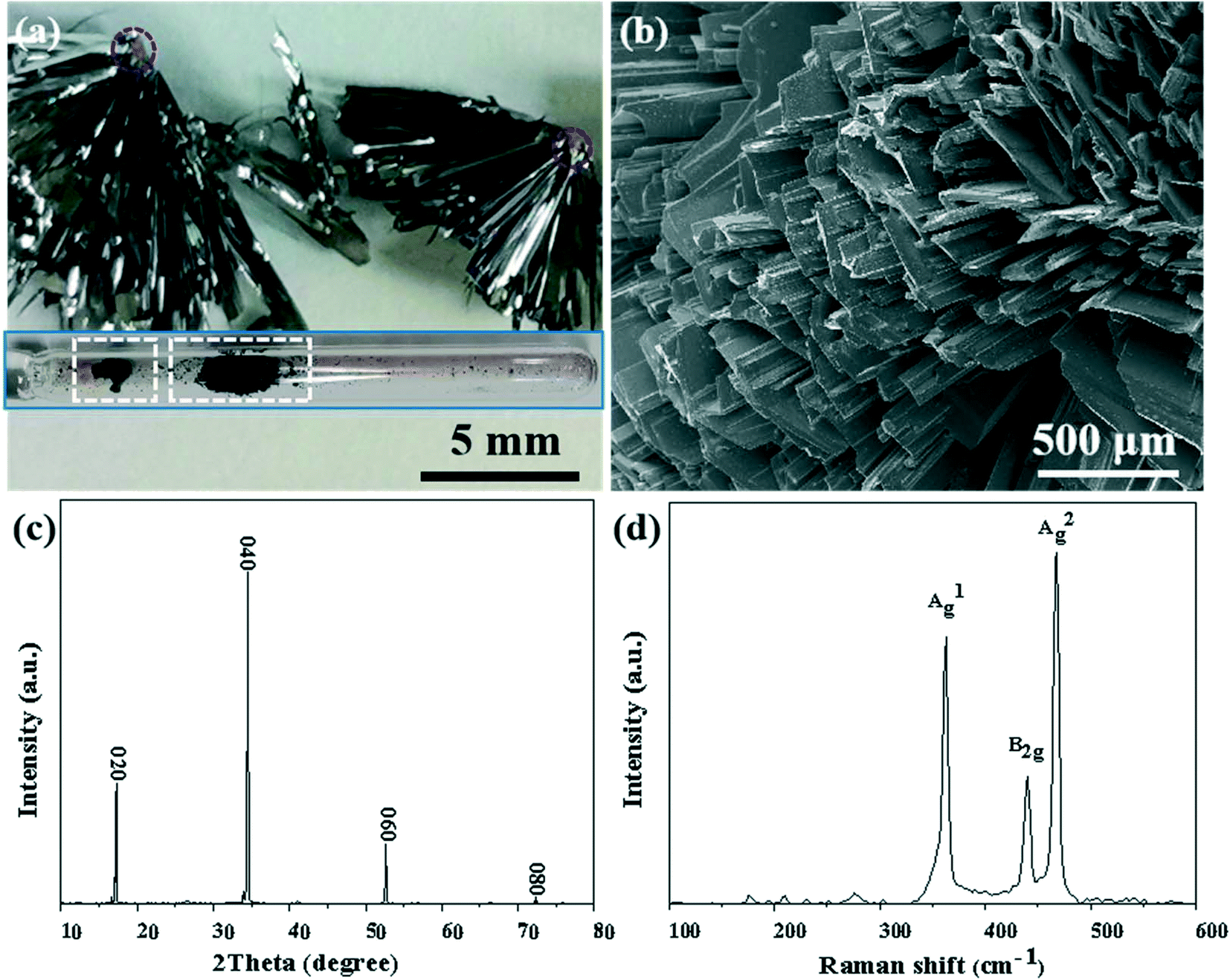
As shown in Fig. 1, BP crystals were successfully synthesized in the aRP–Sn–I2 system of the CVT reaction based on the typical temperature-controlled procedure mentioned in the experimental section. The prepared BP product attached to the wall of the ampoule, and no obvious by-products were observed (the inset of Fig. 1a). However, the cross-sectional view showed that the BP product aggregated on the dark red nucleating agent in the form of ribbons (marked with a red circle). The SEM image reveals that the prepared BP was composed of numerous flat microbelt-like structures (Fig. 1b). Meanwhile, strong sharp peaks with 2theta at 17.0°, 34.3° and 52.4° corresponding to the {020}, {040} and {060} reflections were observed in the X-ray diffraction (XRD) pattern of mashed BP crystals (Fig. 1c), further confirming the layered features of BP. The structural information of BP was further characterized by the Raman technique as shown in Fig. 1d. Three characteristics peaks of BP were observed and can be assigned to A1g (out-of-plane, 361.02 cm−1), B2g (in-plane to zigzag orientation, 438.10 cm−1), and A2g (in-plane to armchair orientation, 466.91 cm−1), which agree well with previously reported results.32 In general, orthorhombic BP crystals, with excellent quality, can be satisfactorily synthesized in the presence of the Sn–I mineralizer under typical reaction conditions.
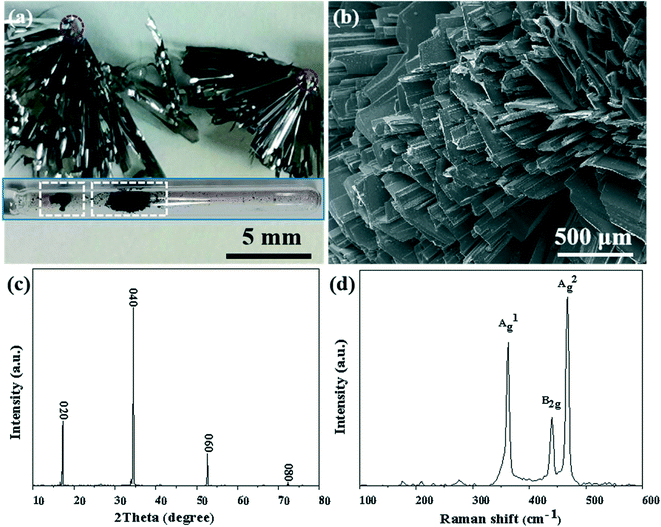 |
| | Fig. 1 (a) Enlarged photograph of BP and nucleating agent (marked with red circle). Inset shows a photograph of the product (marked with white frame) in the ampoule. (b) SEM image of the BP crystal. (c) XRD spectrum of BP. (d) Raman spectrum of the BP crystal. | |
3.1 Characterization and analysis of BP nucleating agent
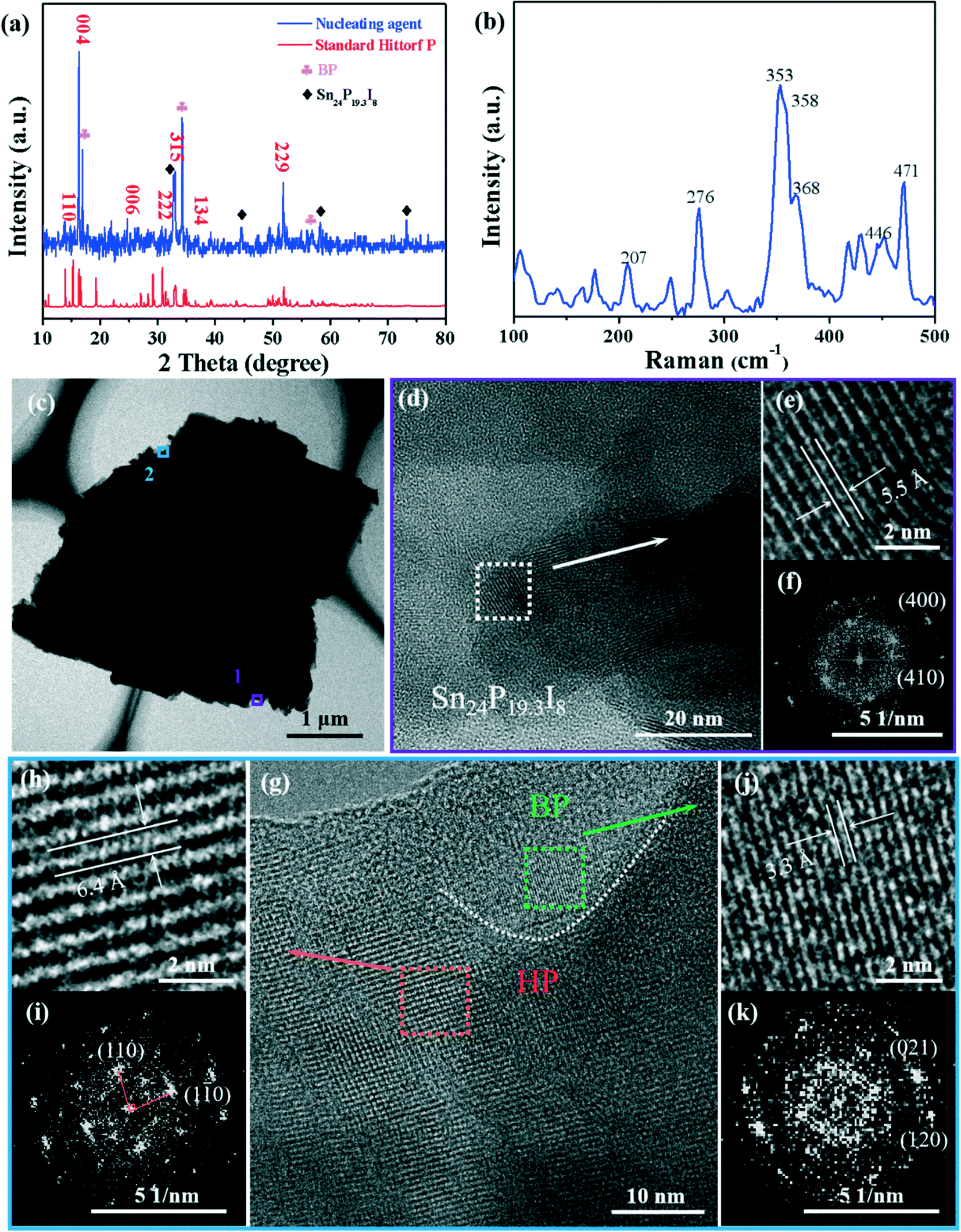
In order to explore the nucleation growth mechanism of the high-yield BP crystals, the dark red nucleating agent (marked by the red circle in Fig. 1a) was characterized and analyzed. The powder XRD peaks of the nucleating agent can be labeled HP (PDF#44-0906) and the peaks marked with a diamond sign were the compound Sn24P19.3I8 (PDF#89-6562) (Fig. 2a). In addition, since the nucleating agent has not been washed with a solution, the diffraction peaks of the BP crystals (PDF#73-1358), labeled with a clover sign, can be observed. Moreover, the Raman spectrum (Fig. 2b) of the nucleating agent shows that the signal peaks of HP are at 207, 276, 353, 358, 368, 446 and 471 cm−1, which are consistent with previous reports.30,33 In addition, the SEM-EDS mapping images of the nucleating agent further proved the existence of the Sn–P–I compound (Fig. S1†).
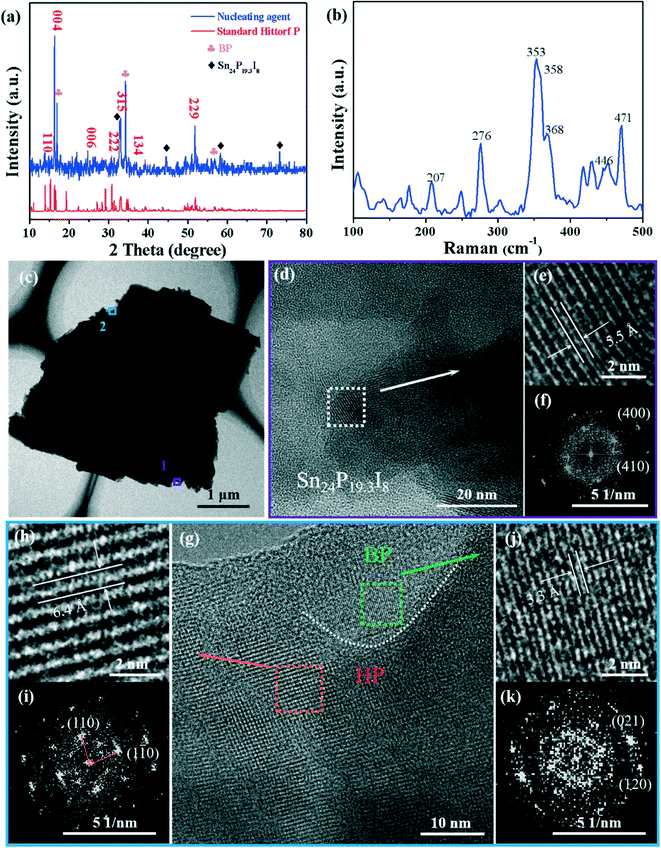 |
| | Fig. 2 Characterization of the BP nucleating agent. (a) XRD pattern of the nucleating agent. (b) Raman spectra of the nucleating agent. (c) TEM image of the nucleating agent. (d) HRTEM image of the nucleating agent taken from the lower edge position 1 in (c). (e) Top right, magnified image of (d). (f) Bottom right, associated fast Fourier transform (FFT) of (e). (g) HRTEM image (middle) of the nucleating agent taken from the upper edge position 2 in (c). (h) Top left, magnified image of the orange area. (i) Bottom left, associated FFT of (h). (j) Top right, magnified image of the green area; (k) bottom right, associated FFT of (j). | |
Furthermore, HRTEM and EDS-mapping further characterized the composition of the nucleating agent. As shown in the HRTEM image in Fig. 2e, the interplanar spacing of 5.5 Å corresponds to the (200) plane of Sn24P19.3I8, which is an enlarged view of Fig. 2d from position 1 in Fig. 2c. The enlarged view (Fig. 2g) of the superstructure of the nucleating agent (Fig. 2c, position 2) shows clear grain boundaries (white dashed lines), and the clear interplanar spacings on both sides of the dashed line are 3.3 Å and 6.4 Å, corresponding to the (021) plane of BP and the (110) plane of HP, respectively.33,34 Therefore, the morphology shown in Fig. 2c from position 1 to position 2 can be described as a sandwich structure formed by the Sn24P19.3I8 compound, HP, and BP. Besides, as can be seen from the STEM-EDS (Fig. S3†) elemental mapping of P, I, and Sn, the elemental distribution is consistent with the above observed structure. This phenomenon is similar to the previously reported HP–BP phase transition growth mechanism,30 but the difference lies in the characterization results of the high-purity growth products.
3.2 Study on the crystallization process of BP
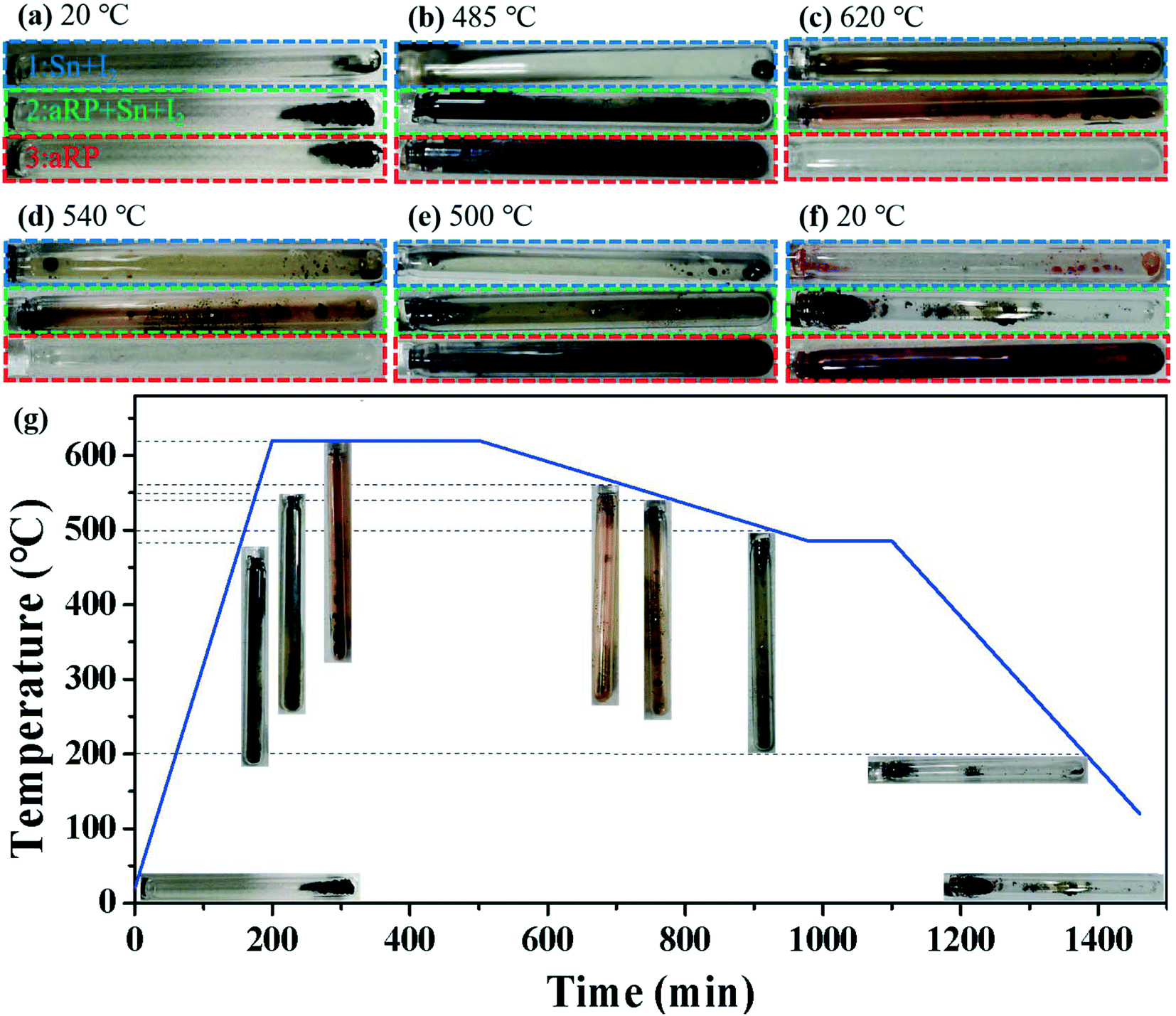
To further study the nucleation process of BP in the CVT reaction, the real-time growth process of synthetic BP was recorded under the classical temperature control program (Fig. 3). The reactants (aRP, Sn, I2) were vacuum sealed in an ampoule labeled 2 for the reaction. As a control experiment, the mineralizer and aRP were individually sealed in ampoules labeled 1 and 3, respectively. As shown in Fig. 3c, orange-red intermediate gas appears in ampoule-2 as the heating process proceeds, and a black liquid is produced at the initial position, which is similar to the mineralization phenomenon in ampoule-1. According to the Clausius–Clapeyron equation, the saturated vapor pressure of tin is 10−7 Pa at 620 °C, which mainly exists as a liquid, and a small amount of gaseous tin and iodine will form compounds.29 During the cooling process, the partial pressure gradient in the ampoule caused the gas to be transported to the low temperature area on the left side of the ampoule,23 and significant deposits were obtained at around 540 °C. As the transport and deposition process continued, a massive BP crystal was formed in ampoule-2 (Fig. 3f). In particular, ampoule-3 had a significant color change around 500 °C (Fig. 3e), and then a red substance attached to the wall of the ampoule was obtained at room temperature. It is worth noting that ampoule-3 did not produce any deposits at 540 °C (Fig. 3d); that is, the crystallization kinetics of aRP were sluggish without the addition of mineralizers, but can be accelerated under the action of mineralizers.
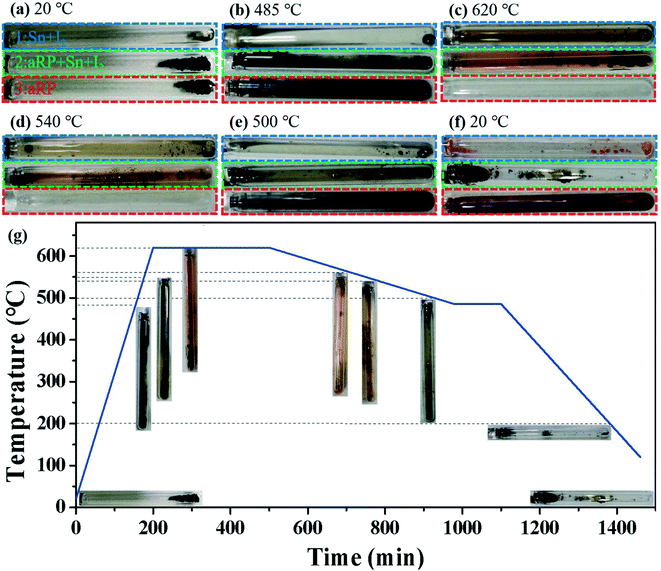 |
| | Fig. 3 (a–f) Optical photos of different temperatures during the synthesis of BP in a typical temperature control program. (g) Typical temperature versus time curve of synthetic BP crystals. | |
3.3 Reaction interrupted experiments to study BP nucleation
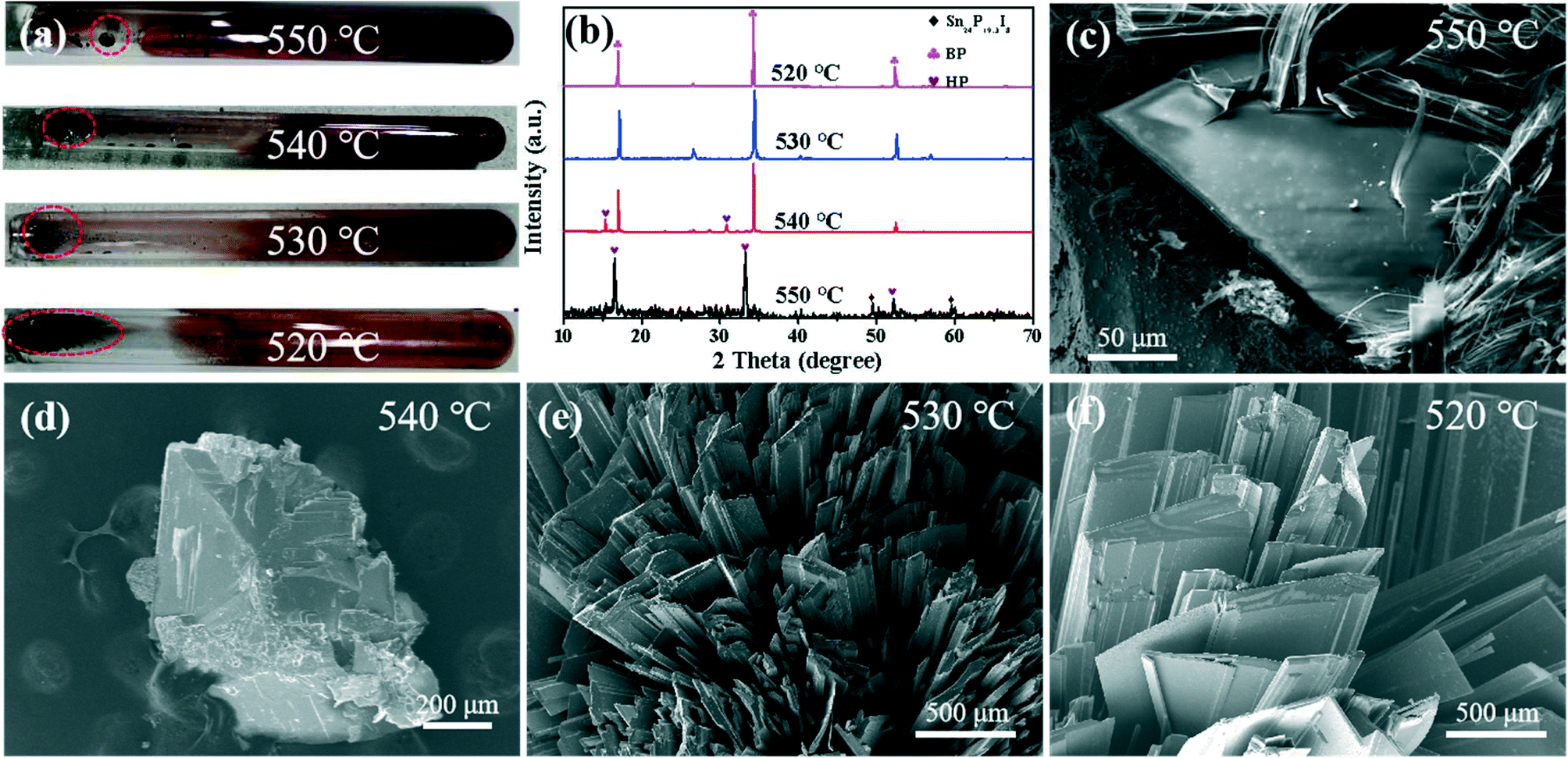
The above experimental results show that the nucleation temperature of BP is about 540 °C. In order to reveal the nucleation mechanism of BP, the intermediate products of the BP nucleation stage were obtained through ex situ reaction interruption experiments. The transportation and deposition process continued along with the drop in reaction temperature. As shown in Fig. 4, the XRD pattern of the irregularly shaped deposits obtained at 550 °C shows that strong HP signals are detected at 16.5°, 33.1°, and 52.1°.33 Among them, the weaker Sn24P19.3I8 signal only exists as a small amount of P–Sn–I compounds at 560 °C (Fig. S3†). Similarly, the elemental distribution diagram shows that the pure P element is distributed on the P–Sn–I ternary compound, which is consistent with the HRTEM results in Fig. 2 above. The SEM images in Fig. 4d–f clearly show that the size of the intermediate product increases with the progress of the reaction in the range of 540–520 °C. Moreover, the XRD pattern (Fig. 4b) shows that the intermediate product at 540–520 °C has the same three peaks at 17.0°, 34.3° and 52.4°, which corresponds well to the peak of BP.32 It is indicated that the phase transition between HP and BP only occurs in the range of 550–540 °C, which is quite different from the previously reported phase transition after HP has grown completely.29,30 The Raman spectrum shows that the gas condensation product on the right side of the ampoule is white phosphorus (WP) (Fig. S5†);38 therefore, the structural transformation of the P4 molecule during the rapid cooling process can almost be ruled out.
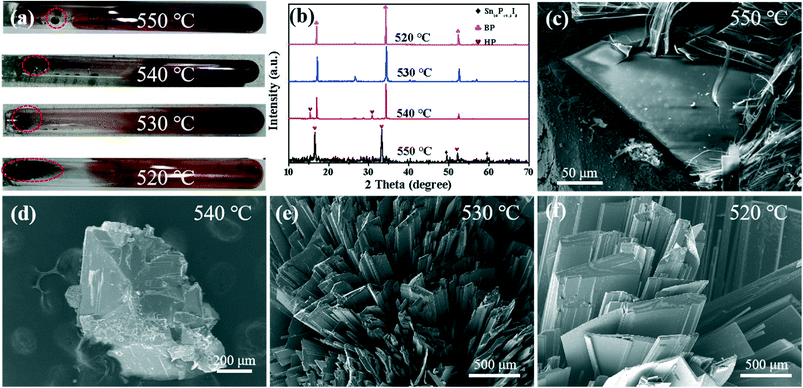 |
| | Fig. 4 (a) Optical photographs of the product obtained at 550–520 °C at room temperature. (b) XRD patterns of the samples at different temperatures marked by the red circle in (a). SEM images marked with red circles in (a) for (c) 550 °C, (d) 540 °C, (e) 530 °C and (f) 520 °C. | |
3.4 The effect of HP nucleation on BP
The reaction rate of 560–540 °C during the deposition phase was adjusted to illustrate the effect of HP nucleation on the growth of BP (Fig. S6†). As shown in Fig. 5a, the layered BP with a larger size of several millimeters was obtained at a growth rate of 0.5 °C min−1, and HP was hardly observed in the larger single crystal. However, the growth of HP was significantly promoted over a longer reaction time. Similarly, compared to the growth rate of 0.3 °C min−1, the BP yields were reduced at both the growth rates of 0.1 and 0.5 °C min−1, especially after increasing the reaction rate. This indicated that HP nucleation played a vital role in the growth of BP. Furthermore, it revealed that HP and BP had a competitive relationship in the nucleation process, and the slow growth rate was conducive to the nucleation and growth of HP at high temperatures (Fig. 5c).31 However, the BP nucleation gradually dominated as the temperature decreased, which promoted the phase transition from HP to BP.
 |
| | Fig. 5 Representative optical photos of black phosphorous products at different cooling rates at 560–540 °C: (a) 0.5 °C min−1 (the inset is a low-magnification optical photo), (b) 0.3 °C min−1, (c) 0.1 °C min−1 (the inset is a partial enlarged view). (d) The SEM image of BP from (c). | |
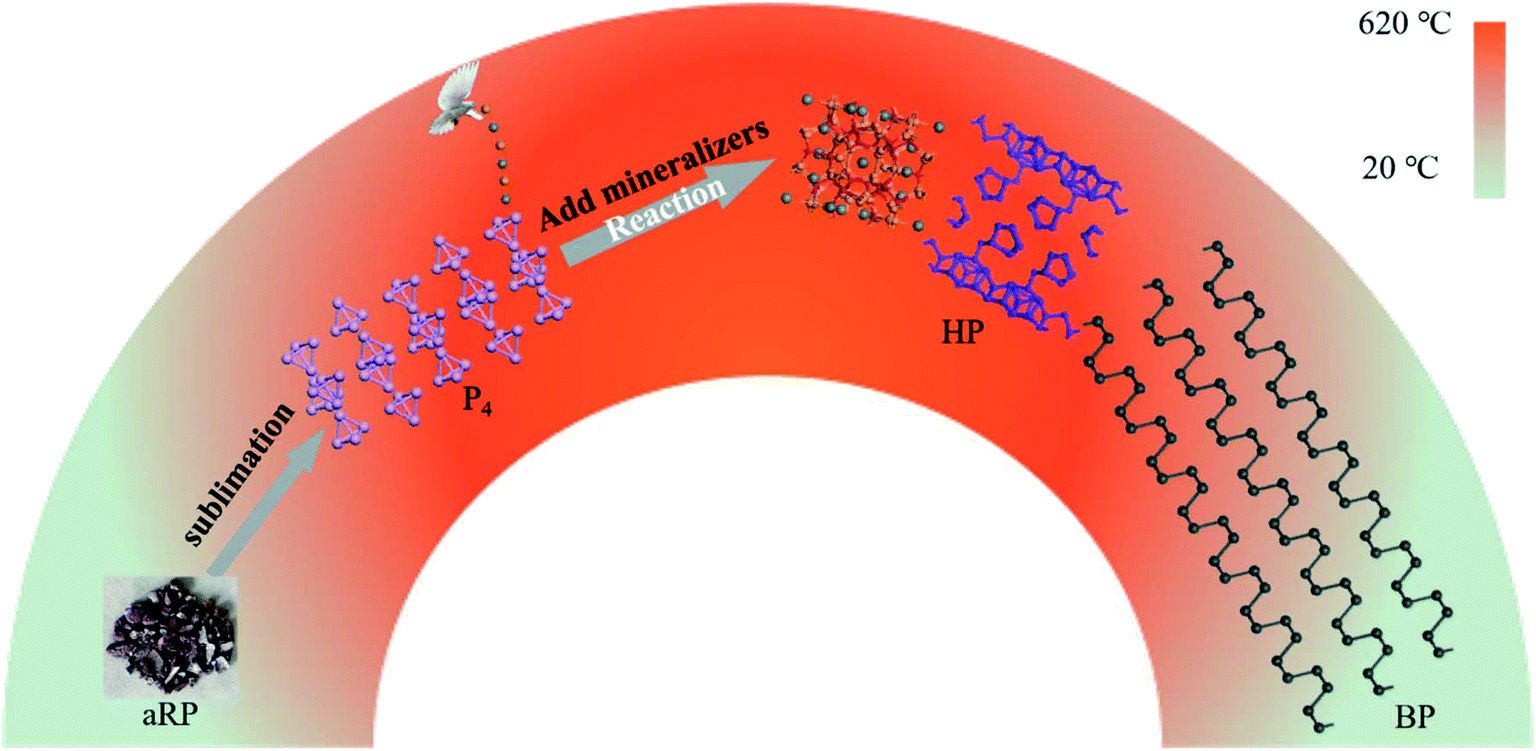
In short, we observed three structural transformations of aRP–P4-HP–BP in the process of synthesizing BP with the aRP–Sn–I system for the CVT reaction (Fig. 6). The first phase transition was from the amorphous structure of RP to the molecular structure of P4, which is reversible, at about 485 °C in the sublimation stage. Subsequently, a small amount of P4 molecules and mineralizers formed P–Sn–I compounds, which may be caused by the further weakening of the bond angle tension between the P orbitals of the P4 molecules themselves under the action of Sn and I.35–37 The existence of P vacancies in the Sn24P19.3I8 compound had become one of the main reasons for promoting the transformation of P4 molecules to the HP structure.28 However, the decrease in temperature caused the transformation of P4 to BP to dominate, while HP growth was inhibited.31 The phase transition between HP and BP was actually a nucleation competition, but HP provided nucleation sites for BP, which is one of the main factors affecting the morphology and yield of BP.
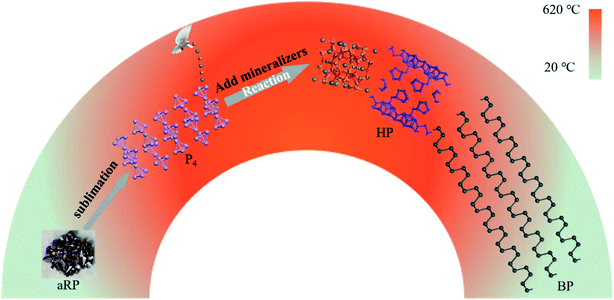 |
| | Fig. 6 Schematic diagram of the transformation of aRP into BP during CVT. | |
4. Conclusions
In summary, the CVT method successfully synthesized high-quality BP crystals in the aRP–Sn–I2 system. We proposed the phase transition process of aRP–P4-HP–BP and revealed the nucleation mechanism of aRP to BP through a series of experiments and characterizations. First, aRP was transformed into a P4 molecular structure at about 485 °C in the sublimation stage. Subsequently, a small amount of P4 molecules were gradually converted into HP at about 560–550 °C during the transportation and deposition stage. However, in the range of 550 to 540 °C, BP nucleation rapidly inhibited the growth of HP and completed the phase transition. As the transportation and deposition processes continued, more P4 molecules were transformed into the BP structure, which promoted the growth of BP crystals. In particular, BP crystals of several millimeters to several micrometers were obtained by regulating the competitive nucleation rate between HP and BP. This provides an idea for the bottom-up preparation of BP nanostructures.
Author contributions
S. Z. performed the material synthesis and TEM/STEM observations, processed the experimental data and drafted the manuscript. Q. L., Y H., Q. K., H. M., and Z. C. helped with nanocrystal synthesis and property tests. H. X., Y. X., B. C., L. S., and B. H. supported the microscopic experiments and data analysis. B. X. and S. M. directed the entire study. All authors participated in discussions and commented on the manuscript.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21972103) and the Key R&D Program of Shanxi Province (201703D111026).
References
- A. Carvalho, M. Wang, X. Zhu, A. S. Rodin, H. Su and A. H. Castro Neto, Nat. Rev. Mater., 2016, 1, 1–16 CrossRef
 .
.
- G. Qin and M. Hu, Small, 2018, 14, 1702465 CrossRef .
- H. Jiang, H. Shi, X. Sun and B. Gao, ACS Photonics, 2018, 5, 2509–2515 CrossRef CAS .
- X. Ling, S. Huang, E. H. Hasdeo, L. Liang, W. M. Parkin, Y. Tatsumi, A. R. Nugraha, A. A. Puretzky, P. M. Das, B. G. Sumpter, D. B. Geohegan, J. Kong, R. Saito, M. Drndic, V. Meunier and M. S. Dresselhaus, Nano Lett., 2016, 16, 2260–2267 CrossRef CAS PubMed .
- N. Mao, J. Tang, L. Xie, J. Wu, B. Han, J. Lin, S. Deng, W. Ji, H. Xu, K. Liu, L. Tong and J. Zhang, J. Am. Chem. Soc., 2016, 138, 300–305 CrossRef CAS PubMed .
- H. Liu, A.-T. Neal, Z. Zhu, Z. Luo, X. Xu, D. Tomanek and P.-D. Ye, ACS Nano, 2014, 8, 4033–4041 CrossRef CAS PubMed .
- R. Fei, A. Faghaninia, R. Soklaski, J. A. Yan, C. Lo and L. Yang, Nano Lett., 2014, 14, 6393–6399 CrossRef CAS PubMed .
- L. Chen, G. Zhou, Z. Liu, X. Ma, J. Chen, Z. Zhang, X. Ma, F. Li, H. M. Cheng and W. Ren, Adv. Mater., 2016, 28, 510–517 CrossRef CAS .
- M. Qiu, Z. T. Sun, D. K. Sang, X. G. Han, H. Zhang and C. M. Niu, Nanoscale, 2017, 9, 13384–13403 RSC .
- J. Pang, A. Bachmatiuk, Y. Yin, B. Trzebicka, L. Zhao, L. Fu, R. G. Mendes, T. Gemming, Z. Liu and M. H. Rummeli, Adv. Energy Mater., 2018, 8, 1702093 CrossRef .
- L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2014, 9, 372–377 CrossRef CAS PubMed .
- R. Kurapati, K. Kostarelos, M. Prato and A. Bianco, Adv. Mater., 2016, 28, 6052–6074 CrossRef CAS PubMed .
- W. Tao, X. Zhu, X. Yu, X. Zeng, Q. Xiao, X. Zhang, X. Ji, X. Wang, J. Shi, H. Zhang and L. Mei, Adv. Mater., 2017, 29, 1603276 CrossRef PubMed .
- L. Qin, S. Jiang, H. He, G. Ling and P. Zhang, J. Controlled Release, 2020, 318, 50–66 CrossRef CAS .
- R. Gusmao, Z. Sofer and M. Pumera, Angew. Chem., Int. Ed., 2017, 56, 8052–8072 CrossRef CAS .
- X. Zhang, J. Deng, J. Yan, Y. Song, Z. Mo, J. Qian, X. Wu, S. Yuan, H. Li and H. Xu, Appl. Surf. Sci., 2019, 490, 117–123 CrossRef CAS .
- B. Li, C. Lai, G. Zeng, D. Huang, L. Qin, M. Zhang, M. Cheng, X. Liu, H. Yi, C. Zhou, F. Huang, S. Liu and Y. Fu, Small, 2019, 15, e1804565 CrossRef PubMed .
- J. C. Jamieson, Science, 1963, 139, 1291–1292 CrossRef CAS PubMed .
- A. Brown and S. Rundqvist, Acta Crystallogr., 1965, 19, 684–685 CrossRef CAS .
- S. H. Aldave, M. N. Yogeesh, W. Zhu, J. Kim, S. S. Sonde, A. P. Nayak and D. Akinwande, 2D Mater., 2016, 3, 014007 CrossRef .
- Q. Xu, Y. Zhu, C. Shi, N. Zhang and T. Xie, J. Mater. Sci.: Mater. Electron., 2020, 31, 19093–19105 CrossRef .
- M. Khurram, Z. Sun, Z. Zhang and Q. Yan, Inorg. Chem. Front., 2020, 7, 2867–2879 RSC .
- D. Wang, F. Luo, M. Lu, X. Xie, L. Huang and W. Huang, Small, 2019, 15, 1804404 CrossRef PubMed .
- D. Wang, P. Yi, L. Wang, L. Zhang, H. Li, M. Lu, X. Xie, L. Huang and W. Huang, Front. Chem., 2019, 7, 21 CrossRef CAS PubMed .
- M. Zhao, H. Qian, X. Niu, W. Wang, L. Guan, J. Sha and Y. Wang, Cryst. Growth Des., 2016, 16, 1096–1103 CrossRef CAS .
- Z. Zhang, X. Xin, Q. Yan, Q. Li, Y. Yang and T.-L. Ren, Sci. China Mater., 2016, 59, 122–134 CrossRef CAS .
-
M. Binnewies, R. Glaum, M. Schmidt and P. Schmidt, Chemical Vapor Transport Reactions, Walter de Gruyter GmbH & Co. KG, Berlin/Boston, 2012 Search PubMed .
- S. Li, X. Liu, X. Fan, Y. Ni, J. Miracle, N. Theodoropoulou, J. Sun, S. Chen, B. Lv and Q. Yu, Cryst. Growth Des., 2017, 17, 6579–6585 CrossRef CAS .
- Z. Chen, Y. Zhu, J. Lei, W. Liu, Y. Xu and P. Feng, CrystEngComm, 2017, 19, 7207–7212 RSC .
- Z. Zhang, D.-H. Xing, J. Li and Q. Yan, CrystEngComm, 2017, 19, 905–909 RSC .
- L. Zhang, M. Gu, L. Li, X. Zhao, C. Fu, T. Liu, X. Xu, Y. Cheng and J. Zhang, Chem. Mater., 2020, 32, 7363–7369 CrossRef CAS .
- S. Sugai and I. Shirotani, Solid State Commun., 1985, 53, 753–755 CrossRef CAS .
- L. Zhang, H. Huang, B. Zhang, M. Gu, D. Zhao, X. Zhao, L. Li, J. Zhou, K. Wu, Y. Cheng and J. Zhang, Angew. Chem., Int. Ed., 2020, 59, 1074–1080 CrossRef CAS PubMed .
- J. Ran, W. Guo, H. Wang, B. Zhu, J. Yu and S. Z. Qiao, Adv. Mater., 2018, 30, 1800128 CrossRef PubMed .
- Q. Liu, X. Zhang, J. Wang, Y. Zhang, S. Bian, Z. Cheng, N. Kang, H. Huang, S. Gu, Y. Wang, D. Liu, P. K. Chu and X. F. Yu, Angew. Chem., Int. Ed., 2020, 59, 14383–14387 CrossRef CAS PubMed .
- P. Shriber, A. Samanta, G. D. Nessim and I. Grinberg, J. Phys. Chem. Lett., 2018, 9, 1759–1764 CrossRef CAS PubMed .
- A. Samanta and I. Grinberg, J. Phys. Chem. C, 2020, 124, 11050–11056 CrossRef CAS .
- H. Östmark, S. Wallin, N. Hore and O. Launila, J. Phys. Chem. C, 2003, 119, 5918–5922 CrossRef .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ce01425k |
|
| This journal is © The Royal Society of Chemistry 2022 |

 *a,
Xiaodong
Hao
*a,
Xiaodong
Hao