
Investigating the solid-state assembly of pharmaceutically-relevant N,N-dimethyl-O-thiocarbamates in the absence of labile hydrogen bonds†
Davin
Tan
*a,
Zi Xuan
Ng
a,
Rakesh
Ganguly
ab,
Yongxin
Li
a,
Han Sen
Soo
 a,
Sharmarke
Mohamed
*c and
Felipe
García
*a
a,
Sharmarke
Mohamed
*c and
Felipe
García
*a
aDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, Singapore 637371, Singapore. E-mail: davin.tan@mail.mcgill.ca; fgarcia@ntu.edu.sg
bChemistry Department, Shiv Nadar University, Gautam Buddha Nagar, 201314 India
cDepartment of Chemistry, Khalifa University of Science and Technology, PO BOX 127788, Abu Dhabi, United Arab Emirates. E-mail: sharmarke.mohamed@ku.ac.ae
First published on 30th October 2020
Abstract
There are many active pharmaceutical ingredients that lack N–H, O–H and S–H hydrogen-bond donor functional groups. N,N-Disubstituted O-thiocarbamates are examples of molecules that display such a feature. Despite the desirable medicinal properties displayed by some N,N-disubstituted O-thiocarbamates, the study of the solid-state properties of these compounds has been relatively unexplored. Herein, we report the synthesis and analysis of the structures and properties of a series of N,N-dimethyl-O-thiocarbamates, and use X-ray diffraction techniques to gain insight into how these molecules self-assemble in the solid-state. As part of our work, we report for the first time the crystal structure of tolnaftate, an active pharmaceutical ingredient that is indicated for the treatment of fungal infections. It was observed that the aryl-thiocarbamate C–O bonds are twisted such that the planar aryl and carbamate moieties are orthogonal. Such a non-planar molecular geometry affects the way the molecules pack and crystal structure analyses revealed four general modes in which the molecules can assemble in the solid-state, with some members of the series displaying isostructural relationships. Computational modelling of the cohesive energy densities in the crystals suggests that there is no single stacking type that is associated with greater stability. However, crystals with a combination of high packing index and π⋯π stacking interactions appear to display large cohesive energy densities. The lack of strong hydrogen bonding interactions in the crystals also leads to relatively low Young's moduli that are within a narrow range of 10–15 GPa for all 14 crystal structures reported.
The ability to predict and dictate how molecules assemble in the solid-state has been the main cornerstone of crystal engineering.1–5 Creating reproducible intermolecular interactions is thus crucial as it can be used as a design element to make different types of crystalline solids and materials, such as energetic compounds,6,7 three-dimensional framework structures,8,9 and cocrystals.10–14 Formation of such supramolecular synthons15,16 enables solid-state chemists to engineer how molecules assemble in the solid-state, and this is particularly essential in the context of pharmaceutical solids. Controlling how the active pharmaceutical ingredients (APIs) pack in the solid-state would affect their physicochemical properties such as solubility, compressibility, and dissolution properties, which can in turn be fine-tuned.17–20
Most crystal engineering strategies, such as cocrystallization, typically depend on the construction of hydrogen bonding (HB) interactions using functional groups that contain labile or acidic protons such as amides, amines, alcohols, and carboxylic acids.21–23 However, in many pharmaceutical molecules, such HB donor functional groups are not present, such as diazepam, midazolam, progesterone, and tamoxifen (Fig. 1). In these cases, other types of intermolecular interactions may dominate and affect their assembly in the solid-state. These types of non-hydrogen bonding interactions include halogen bonds, π⋯π or CH⋯π interactions, and van der Waals forces.24–26 One class of biologically active compounds that typically do not contain HB motifs is O-thiocarbamates. These compounds are pharmaceutically-relevant and are known to exhibit antibacterial and anti-fungal properties in marketed drugs such as tolnaftate, tolciclate, and goitrin (Fig. 1).27–29
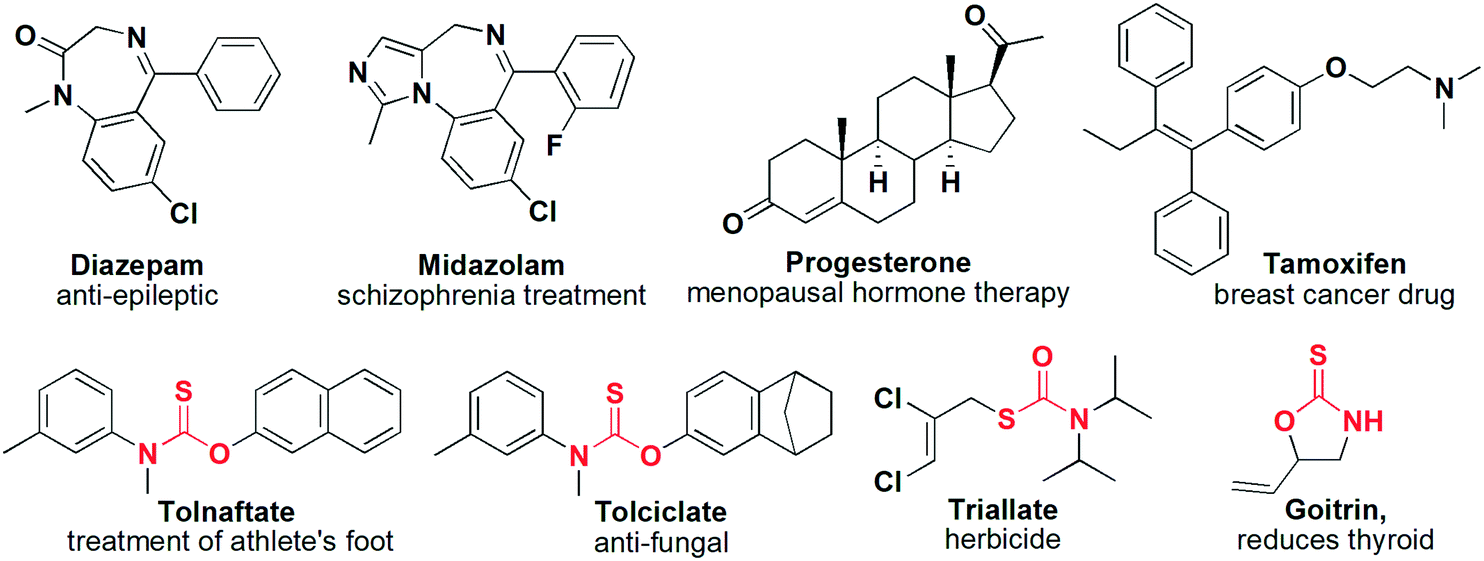 | ||
| Fig. 1 Examples of non-hydrogen bond donor drugs, including thiocarbamate containing pharmaceutical compounds. | ||
However, a quick survey of the Cambridge Structural Database (CSD) revealed that approximately 290 metal-free organic compounds containing the O-thiocarbamate functional group have been reported.30 This number dwindles to 91 for compounds that contain N,N-disubstituted O-thiocarbamates, in which the carbamate disubstituted N atoms act as HB acceptors. All these compounds containing O-thiocarbamate groups are isolated cases, except for a selected few. Specifically, Tiekink31 and Kimber32 have shown how unsubstituted O-thiocarbamates can be used as supramolecular synthons by forming dimeric pairs or linear chains held by N–H⋯S HB interactions. However, to the best of our knowledge, there has not been any study on understanding how N,N-disubstituted O-thiocarbamate molecules pack and assemble in the solid-state or what their intermolecular interactions are.
In addition, despite biological studies on the activity of tolnaftate, a well-known drug for the treatment of athlete's foot,27 its solid-state structure has not been reported yet. The lack of reports on the crystal structures of these pharmaceutically relevant compounds limits the understanding of how these molecules self-assemble in the solid-state in the absence of intermolecular HBs.
Hence, we set out to synthesize and characterize, using single-crystal X-ray diffraction (SCXRD), a series of related single-component N,N-dimethylaryl-O-thiocarbamates (Fig. 2). These O-thiocarbamates were carefully chosen whereby the substituents on the aryl rings contained a variety of functional groups, including electron withdrawing (2, 3, 6, 10, 11, 12), electron donating (4, 5, 9), and sterically hindered groups via rational modification at the ortho position of the aryl ring (7, 8), a polyaromatic system (13), as well as a N,N-diphenyl compound (14), and the commercial drug tolnaftate (15).
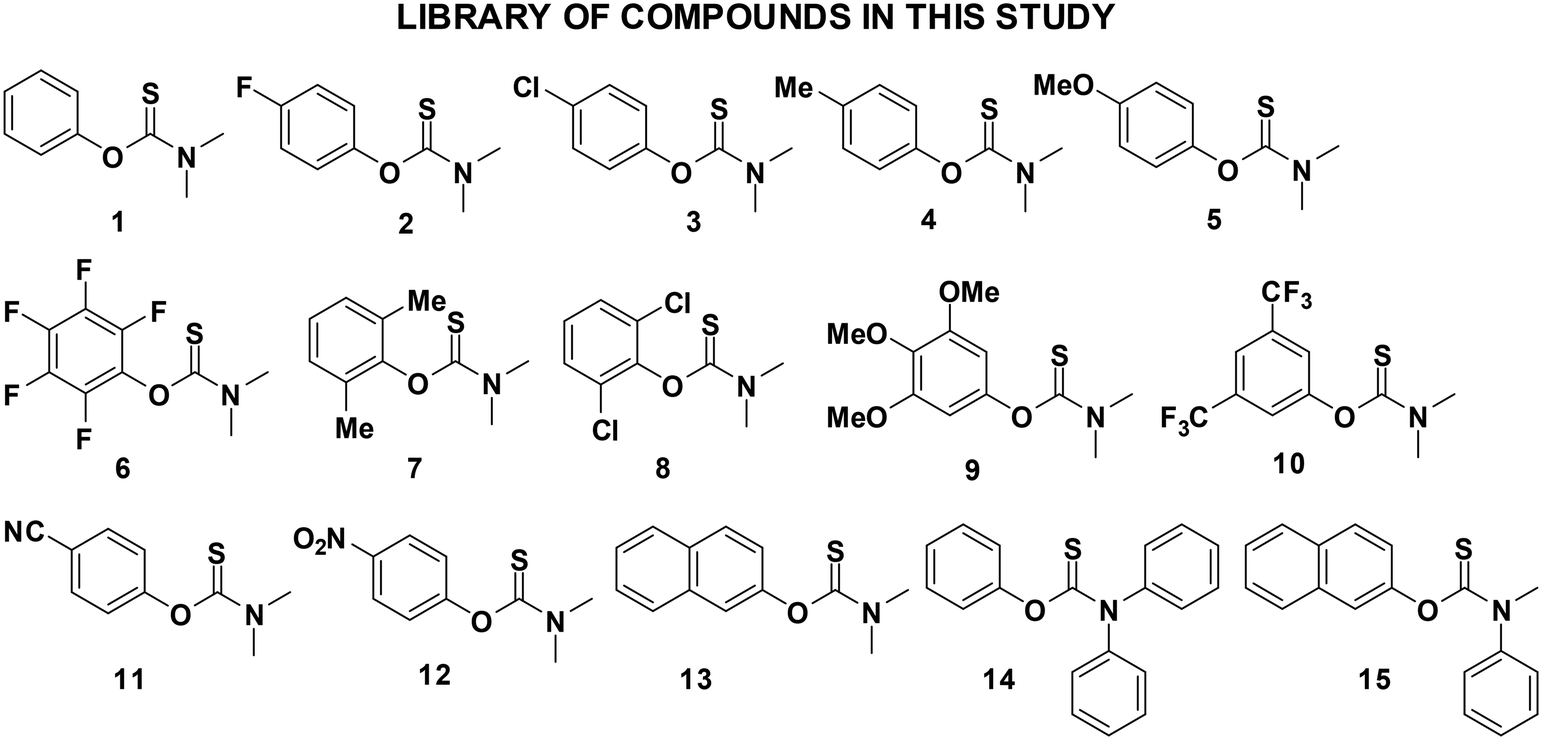 | ||
| Fig. 2 Various N,N-disubstituted O-thiocarbamates investigated. | ||
From a structural standpoint, it may be assumed that thiocarbamates adopt a flat conformation. Such a flat molecular geometry is commonly seen in phenols, carbonates, anilines, thioureas, or ureas,33 where the aryl rings are coplanar with their respective functional group. However, based on the SCXRD data analysis, the molecular structure found in O-thiocarbamate species is far from being flat. Instead, the aryl–O bond is twisted such that the thiocarbamate moiety is almost perpendicular to the aryl ring. This orientation can be attributed to stereoelectronic factors since twisting of the aryl–O bond allows for delocalization and overlap between the two lone pairs of electrons on the O atom with the π electrons of the aromatic ring. Furthermore, the orthogonality between the thiocarbamate group and the aryl ring allows for the S atom to be oriented close to, and directly above, the ipso C atom of the aryl ring. This might explain the propensity of O-thiocarbamates to undergo isomerization, where the S and O atom exchange positions to form S-thiocarbamates under catalytic conditions or at elevated temperatures.34,35
The molecular structure of the studied N,N-disubstituted-aryl-O-thiocarbamates (i.e., compounds 1–15) can be described as two distinct planar groups that are perpendicular to each other, but are connected by an O atom (Fig. 3a). One group (Fig. 3a, in blue) represents the planar thiocarbamate moiety, while the other planar group (Fig. 3a, in green) is the phenol moiety. The dihedral torsional angles α and β describe the twisting of the aryl C–O and the thiocarbamate C–O bonds, respectively. In the absence of directional HBs, it is reasonable to assume that these N,N-dimethylaryl-O-thiocarbamates would adopt similar packing motifs, since the overall geometrical shape, polarity, volume, and size of the compounds do not seem to differ significantly. It is also expected that the molecules should be primarily held together by weak van der Waals or π⋯π interactions in the solid-state.
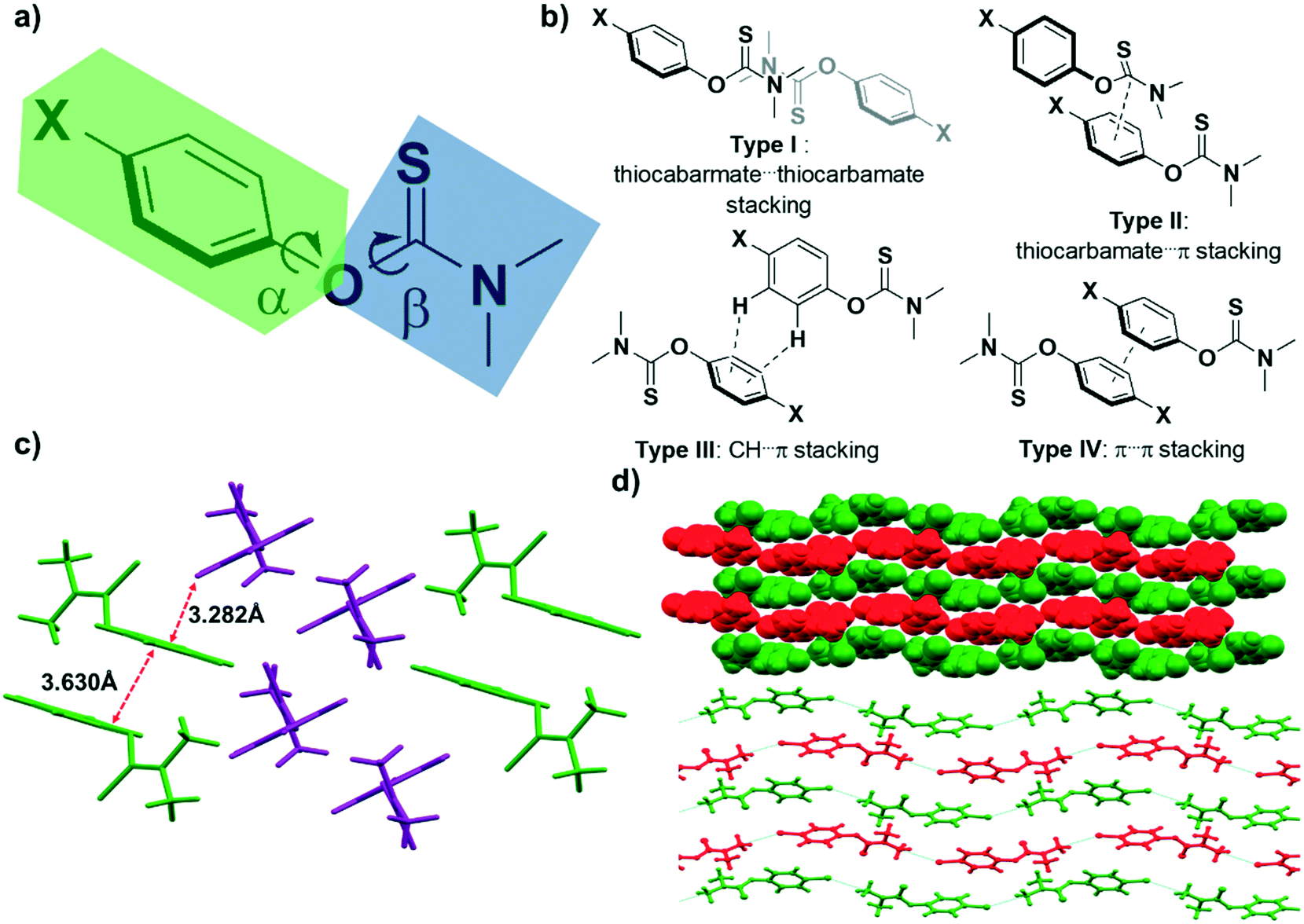 | ||
| Fig. 3 a) Generic structure of N,N-dimethyl-O-thiocarbamate, illustrating the orthogonal thiocarbamate plane (in blue) and the substituted aryl plane (in green). b) The four intermolecular interactions observed from the library of compounds studies, namely type I (thiocarbamate⋯thiocarbamate stacking in an anti-fashion), type II (thiocarbamate–aryl stacking), type III (CH⋯π stacking), and type IV (π⋯π stacking). c) Section of the crystal packing of 2, denoting the type III stacking between two layers and the type IV stacking within each layer. d) Space-filling and ball-and-stick representations of the isostructural molecular packing of 3 and 4 as viewed along the crystallographic bc plane. The two different layers are denoted in red and green. | ||
Our initial attempts to grow diffraction quality crystals of 1 were not successful, yielding only oils or thin opaque films. Fortunately, SCXRD data were obtained for compounds 2–15, and preliminary analyses of their crystal structures revealed four distinct types of packing interactions (Fig. 3b), namely, (i) type I (thiocarbamate⋯thiocarbamate in an anti-fashion), (ii) type II (thiocarbamate–aryl), (iii) type III (CH⋯π), and (iv) type IV (π⋯π) interactions. Subsequently, more detailed analyses were conducted to discern any patterns between the molecular packing of the various compounds. Key interatomic distances and angles are summarized in Table 1. Notably, the aryl C–O and ipso C–S bond distances, as well as the C–O–C angle (the angle between the aryl and thiocarbamate planes) and O–C–S angles did not show significant variation between the different compounds. However, there is a substantial difference in their dihedral angles, 73.72–119.26° and 0.18–22.91°, for α and β, respectively.
| Compound | Type of stacking | Dihedral angle α (°) | Dihedral angle β (°) | C–O–C angle (°) | O–C–S angle (°) | Ipso C–S distance (Å) | PIa (%) | CEDb (MPa) | Young's modulusc (GPa) |
|---|---|---|---|---|---|---|---|---|---|
| a PI is the packing index of the crystal expressed as a %. b CED is the cohesive energy density for the crystal. c The Hill averaging scheme was used to calculate the Young's moduli. | |||||||||
| 2 | III + IV | 89.66, 97.51 | 0.21, 9.90 | 119.24, 120.83 | 124.19, 123.84 | 2.970, 2.999 | 65.80 | 2586 | 13.39 |
| 3 | I + II | 101.35 | 5.71 | 119.36 | 123.45 | 2.972 | 68.00 | 2421 | 12.04 |
| 4 | I + II | 87.67 | 0.58 | 119.05 | 123.36 | 2.961 | 67.60 | 1685 | 10.04 |
| 5 | II | 92.62 | 1.82 | 118.66 | 123.84 | 2.964 | 68.80 | 2449 | 9.84 |
| 6 | I + IV | 95.22 | 5.45 | 118.26 | 122.38 | 2.924 | 67.60 | 2587 | 12.39 |
| 7 | II + IV | 84.88, 94.83 | 11.04, 3.34 | 119.83, 119.43 | 123.26, 123.68 | 2.994, 2.980 | 68.80 | 2081 | 12.12 |
| 8 | IV | 73.86 | 28.71 | 118.19 | 122.41 | 2.997 | 70.60 | 1766 | 12.15 |
| 9 | I | 99.78 | 0.18 | 120.81 | 124.39 | 3.017 | 70.60 | 1933 | 10.56 |
| 10 | III + CF3-π | 123.25 | 18.41 | 119.75 | 123.5 | 3.011 | 62.00 | 3968 | 9.99 |
| 11 | I + II | 107.3 | 20.11 | 117.33 | 122.95 | 2.958 | 70.60 | 3406 | 12.32 |
| 12 | IV | 103.92 | 2.1 | 121.86 | 123.36 | 3.016 | 71.70 | 4253 | 14.86 |
| 13 | III | 119.26 | 3.05 | 119.94 | 124.18 | 2.991 | 70.30 | 1570 | 12.90 |
| 14 | III + IV | 73.72, 79.86, 103.34, 104.54 | 22.91, 20.10, 13.73, 15.73 | 119.43, 118.39, 119.62, 120.76 | 124.03, 124.04, 123.41, 124.60 | 3.022, 2.993, 2.989, 3.035 | 69.40 | 1496 | 11.20 |
| 15 | III + IV | 95.81 | 5.1 | 119.05 | 124.84 | 2.985 | 68.80 | 1595 | 13.21 |
Compound 2 crystallizes in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group and self-assembles forming alternating layers (Fig. 3c). Each layer is held together by type IV stacking interactions, whereas different layers display type III interactions between them. Molecules of 3, on the other hand, do not pack in the same manner as 2 despite having grown in the same solvent (i.e., diethyl ether). Instead, 3 forms type I and II stacking interactions producing relatively planar layers (Fig. 3d), where alternating layers propagate in opposite directions.
space group and self-assembles forming alternating layers (Fig. 3c). Each layer is held together by type IV stacking interactions, whereas different layers display type III interactions between them. Molecules of 3, on the other hand, do not pack in the same manner as 2 despite having grown in the same solvent (i.e., diethyl ether). Instead, 3 forms type I and II stacking interactions producing relatively planar layers (Fig. 3d), where alternating layers propagate in opposite directions.
According to Kitaigorodskii's principle of close packing, molecules with similar functional groups, shape, and size tend to adopt similar packing assemblies and are thus considered isostructural.36 One good example of this is the chloro–methyl exchange rule,37 whereby Cl and Me in related compounds can be isostructural and interchangeable within frameworks with similar crystal packing. Such isostructurality trends also persist within our sample group of O-thiocarbamate species. In particular, 3 and 4 obey the aforementioned chloro–methyl exchange rule. Both compounds crystallize in the same monoclinic P21/c space group, have almost identical unit cell parameters, form similar type I and II stacking interactions, and arrange in the same fashion (i.e., they are isomorphous) (Fig. 3d).38
In 5, the presence of a methoxy group causes significant changes to the crystal structure. These methoxy substituents undergo weak intermolecular C–H⋯O HB interactions with adjacent methoxy groups, forming dimeric pairs. These pairs then form alternating interlocked grids (Fig. S31†) consisting of type II interactions.
The crystal packing for 6 is also unique among our series of compounds. XRD analysis revealed that all the pentafluoroaryl rings within the crystal structure are facing the same direction and are slip-stacked in a type IV manner, along the crystallographic a-axis (Fig. 4a), while the thiocarbamate groups are all type I stacked facing the b-axis. When viewed along the c-axis (Fig. 4b), it becomes clear that all the molecules assemble in the same manner and the thiocarbamate and aryl groups are almost perpendicular (α = 95.22°).
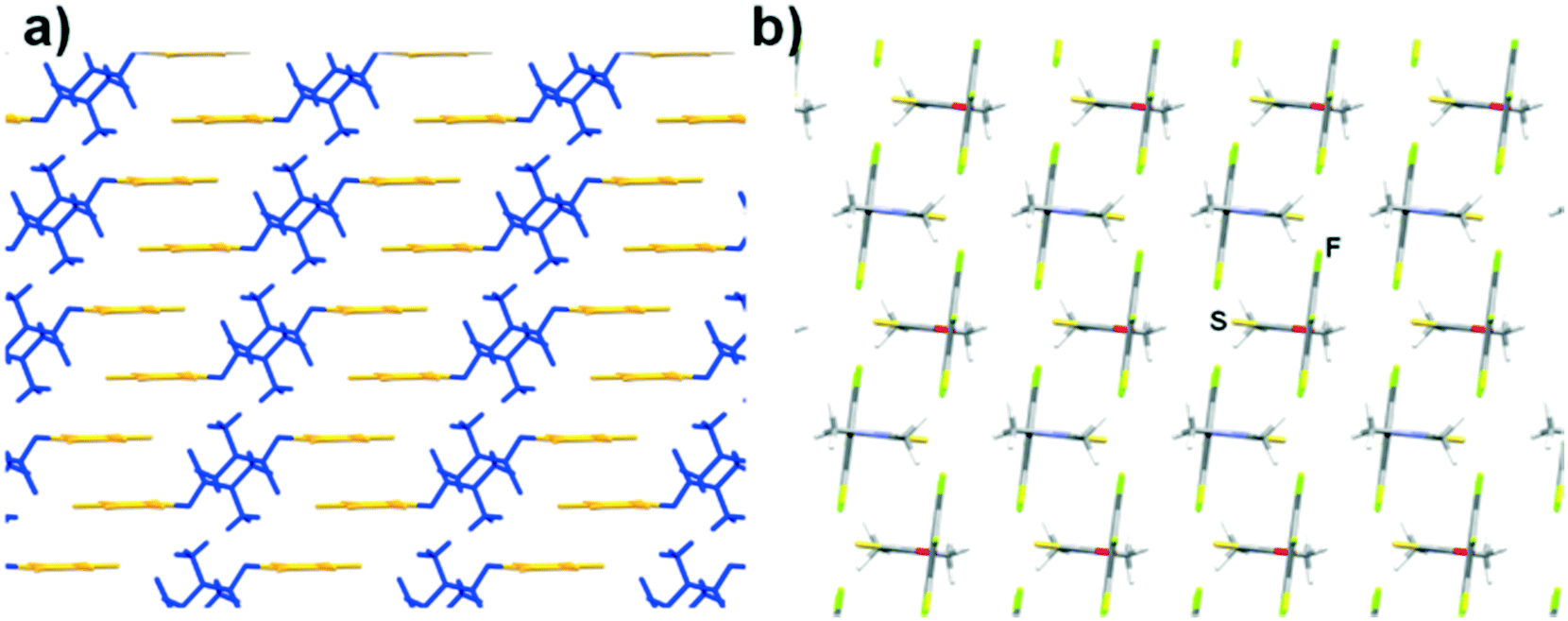 | ||
| Fig. 4 Capped sticks representation of the crystal structure of 6, (a) where the pentafluorophenyl group is highlighted in yellow and the thiocarbamate moiety is coloured in blue, as viewed along the a-axis (a) and along the c-axis (b). | ||
The crystal structure for compound 7 has been previously reported (CCDC code RAHLUK).39 Despite the isostructurality observed in 3 and 4, the chloro–methyl exchange rule was not obeyed in 7 and 8. The aryl rings of compound 7 form type II and IV interactions with one another. However, these interactions are slightly slip-stacked due to the presence of the Me groups at the 2′ and 6′ positions. These molecules then pack to form layers, in which molecules in alternating layers are rotated by 90 degrees (Fig. 5a). Conversely, in 8, the aryl groups only undergo type IV stacking interactions (inter-aryl centroid–centroid distance is 4.023 Å). In addition, the thiocarbamate moieties are aligned in a head-to-tail fashion, forming anti-parallel catemers linked by weak intermolecular S⋯H3CN interactions (Fig. 5b).
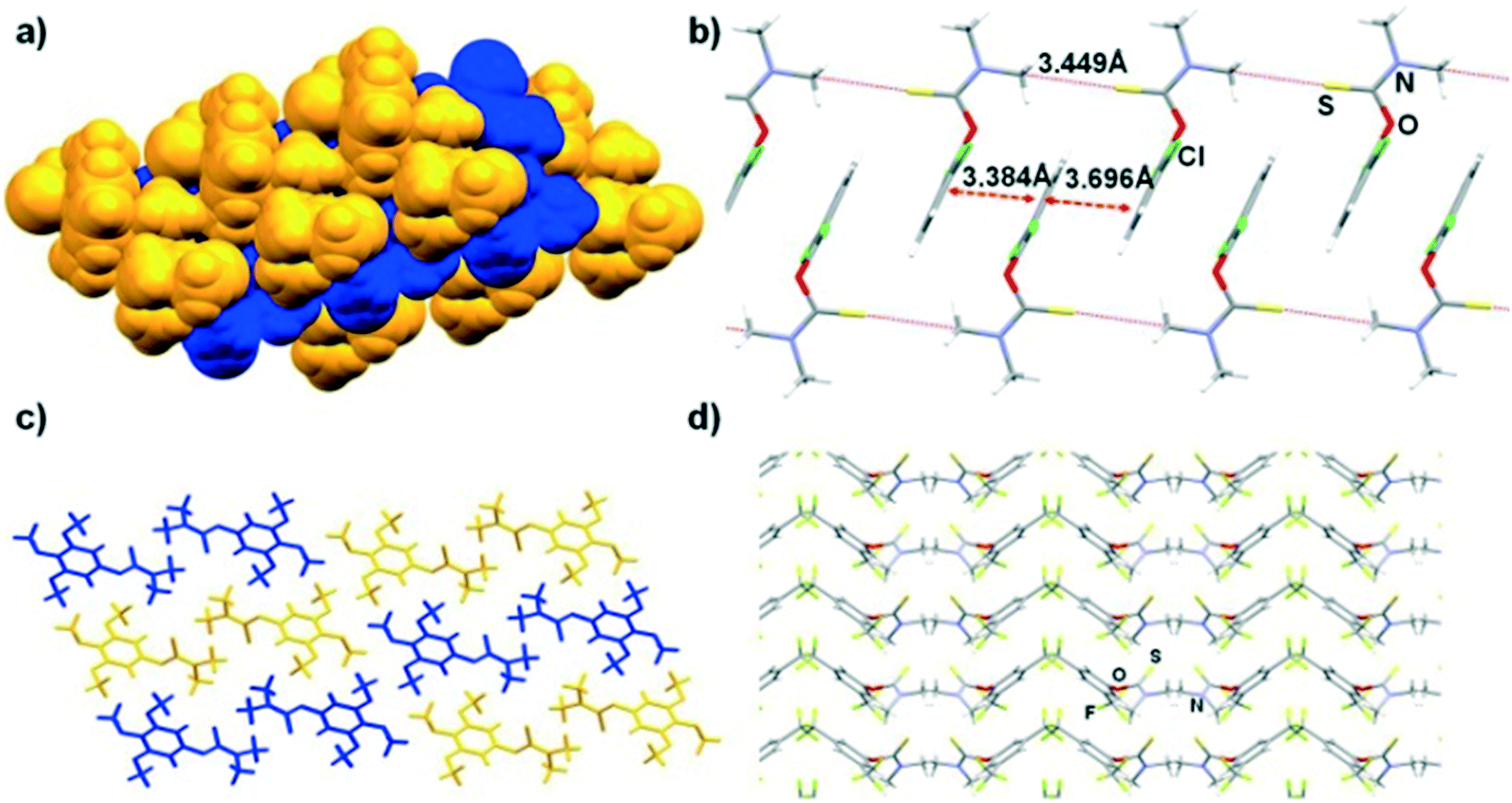 | ||
| Fig. 5 a) Space-filling representation of the crystal structure of 7, denoting two distinct alternating layers in blue and orange. b) Capped stick representation of 8, illustrating type IV stacking interactions between the phenyl moieties. The thiocarbamate moieties of adjacent molecules are aligned into chains. c) Alternating dimeric molecules of 9 associated by type I stacking are shown in yellow and blue. d) Fragment of the crystal structure of 10, denoting the zig-zag wavy packing, propagating along the b-axis. | ||
Similar to 5, the molecules of 9 undergo weak intermolecular C–H⋯O HB interactions, forming an extensive network. However, unlike 5, where the dimers are associated via tail-to-tail pairing of the methoxy groups, the molecules in 9 form dimeric pairs in which the planar thiocarbamate moieties are stacked (type I), and alternating and adjacent pairs of molecules are rotated by 90° (Fig. 5c).
Notably, molecular self-assembly for 10 does not seem to conform to any motif that was previously observed. Whereas the thiocarbamate and aryl moieties display weak intermolecular interactions in 2–9, for 10, neither the thiocarbamate nor the 3,5-bis(trifluoromethyl)aryl moieties are aligned or stacked parallel. Instead, the molecules of 10 form symmetrically associated pairs that are mirror-images. These pairs are arranged in a zig-zag manner forming parallel corrugated chains that propagate along the b-axis (Fig. 5d). In addition, the S atom in the thiocarbamate functional group also acts as a HB acceptor to the slightly acidic ortho C–H of the aryl ring (with a S⋯CF3 distance of 3.736 Å), as well as a combination of type III and CF3⋯ π interaction.
In the solid-state structure of 11, the molecules are arranged in a similar way to 6, where the molecules form pairs in which the thiocarbamate moieties are slipped-stacked in an anti-fashion (type I). These pairs of molecules assemble and form alternating layers that are opposite one another. However, unlike 6 where the aryl groups are parallel and face the same direction allowing for π⋯π stacking between the layers, in 11, the aryl groups are almost perpendicular. Colour coding (Fig. 6a) the thiocarbamate and the 4-cyanoaryl moieties (in purple and cyan, respectively) shows how the perpendicular aryl groups display a linear corrugated arrangement along the crystallographic a-axis. In addition, the NCH3 groups of adjacent molecules are positioned directly above the slightly electron-deficient aryl rings, allowing for type II interactions to occur.
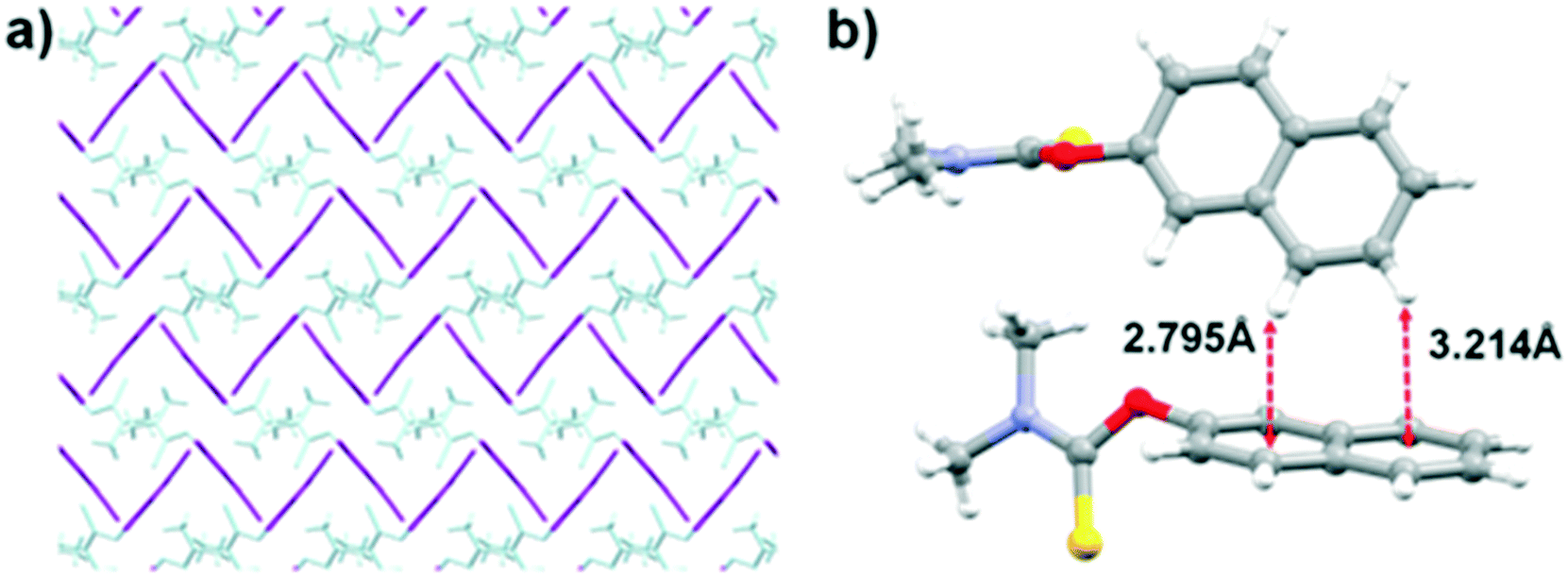 | ||
| Fig. 6 a) Capped stick representation of the crystal structure of 11, clearly showing the corrugation of the aryl groups (in purple) along the a-axis. b) Ball and stick representation of two molecules of 13, illustrating the intermolecular C–H⋯π interactions. | ||
For 12, the molecular packing motif is different from the rest of the compounds. In particular, the presence of highly polar NO2 groups allows for weak CH⋯O interactions to form between the nitro group and NCH3 groups of neighbouring molecules (Fig. S32†). The molecules are also arranged in ways where the aryl moieties are slipped-stacked (type IV, aryl–aryl distance ca. 3.428–3.503 Å) with alternating molecules rotated approximately 120°. These, in turn, form layers that propagate along the a-axis, with adjacent layers aligned in the opposite direction.
It was anticipated that the molecules of 13 would self-assemble in a similar fashion to compound 8, where the thiocarbamates align head-to-tail forming catemeric chains with the naphthyl groups to create an extensive type IV π⋯π stacked system. However, XRD analysis revealed that the molecules of 13 are, instead, aligned such that the thiocarbamate moieties of adjacent molecules are perpendicular. These, in turn, orientate their naphthyl rings above each other, allowing for an extensive network of type III CH⋯π interactions to be formed (Fig. 6b).
In contrast to previous compounds where the N,N-dimethyl groups are small and coplanar to the thiocarbamate moiety, 14 comprises two bulky N,N-diphenyl rings. In this compound, the two bulky N,N-diphenyl rings are perpendicular to the thiocarbamate motif, giving rise to a Y-shaped molecule. In this case, the three bulky non-polar phenyl rings “shield” the more polar thiocarbamate functional group (Fig. 7a). Without the presence of strong HB motifs or exposed polar functional groups, these molecules assemble and pack via the formation of type III and IV interactions. The type IV interactions are only formed between the O-phenyl rings and not with the N-phenyl rings of adjacent molecules. Also, type III interactions are formed between the O-phenyl-to-N-phenyl and N-phenyl-to-N-phenyl rings. Unlike 8, the type IV stacking does not propagate extensively throughout the crystal structure and is only observed between neighbouring pairs of molecules. Crystal packing analysis also revealed that the Y-shaped molecules of 14 form intercalated wave-like layers, in which alternating layers propagate in opposite directions (Fig. 7b). Such a similar wave-like packing motif was also observed in 3.
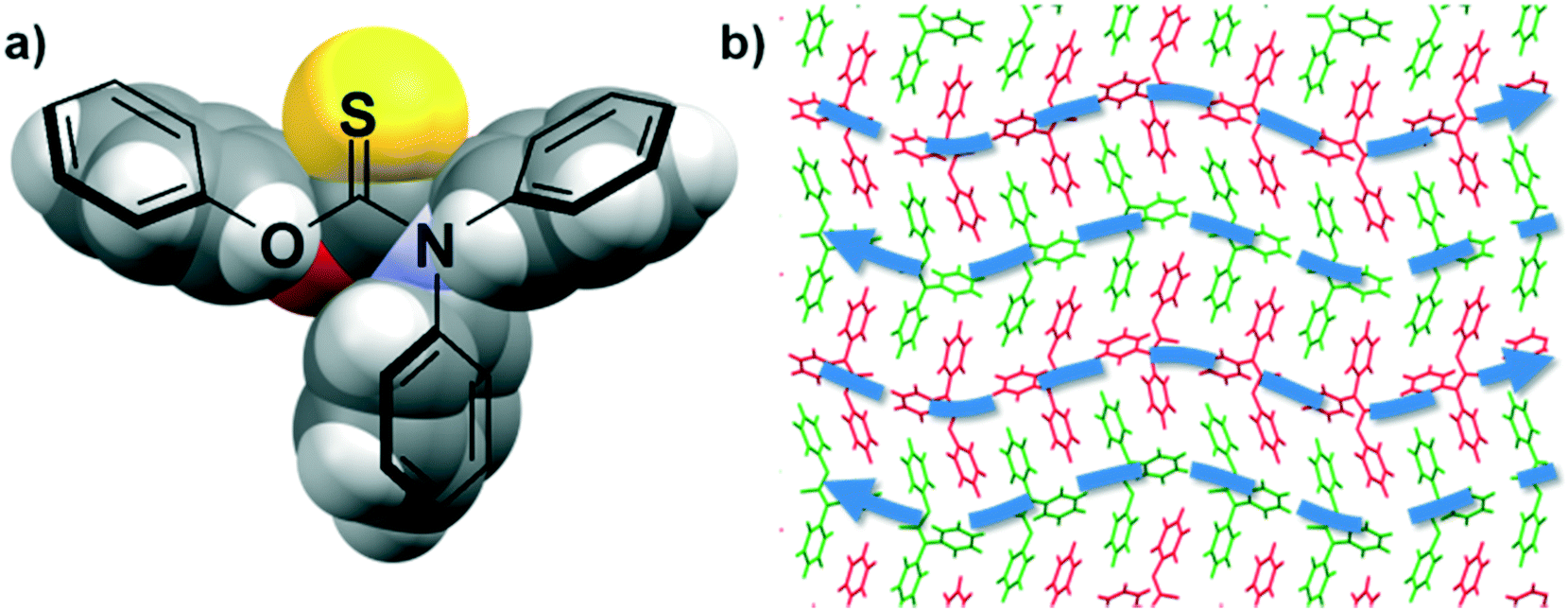 | ||
| Fig. 7 a) Space-filling representation of a Y-shaped molecule of 14. b) Fragment of the crystal structure of 14, showing alternating layers (in red and green) consisting of π⋯π stacking interactions. Blue arrows are drawn to depict the alternating layers propagating in opposite directions. | ||
Lastly, X-ray diffraction quality crystals of 15 (i.e., the anti-fungal drug tolnaftate) were obtained after slow evaporation (three days) of the compound in an acetone and chloroform mixture. Despite having a similar Y-shaped geometry to 14, molecules of 15 instead self-assembled to form inversely associated pairs that resemble the molecular packing of 13, mainly consisting of type IV stacking interactions (aryl–aryl distance ca. 3.551 Å) between their naphthyl rings (Fig. S33†). Additionally, type III interactions (2.832–3.083 Å) between neighbouring O-naphthyl (in yellow) and between adjacent N-phenyl (in blue) rings of adjacent molecules can also be observed.
Computational modelling of the packing indices, cohesive energy densities and mechanical properties of all 14 crystal structures discovered in this work (see Table 1) has revealed a few interesting observations. With the exception of crystal 10, all the reported crystal structures are efficiently packed, with packing indices ranging from 65–72%. The cohesive energy density (CED) is defined as the energy required to separate a unit volume of molecules in the crystal to infinite separation and is therefore a good measure of the strength of the various intermolecular forces in the crystals. In general, we find that there is no obvious correlation between the stacking type (Fig. 2b) and the strength of the intermolecular forces observed in the crystal with many crystals displaying comparable CEDs (Fig. 8a). In fact, the three crystals with the highest CED (11, 10 and 12, respectively) all display different stacking modes. Crystals of 12 display the highest packing index (71.7%), largest Young's modulus and highest CED of all the structures reported, reflecting the efficiently packed thiocarbamate molecules that are sustained by type IV π⋯π stacking interactions with centroid–centroid distances less than 3.7 Å (Fig. 8b). By contrast, crystals of 14 display the lowest CED even though the molecules are sustained via a combination of type III and IV stacking interactions. This is probably because the crystal structure of 14 has four symmetry independent molecules, each of which has three aromatic rings but only one aromatic ring in each molecule engages in type IV stacking interactions (Fig. 8b) as a result of the Y-shaped structure of 14 (Fig. 7a). Thus, crystals of 14 do not display efficient type IV stacking interactions when compared to 12, leading to 14 having a lower CED. Given the lack of strong hydrogen bonding interactions, it is not surprising that the computed Young's moduli (see Table 1) of the crystals are relatively low and all lie within a narrow range of 10–15 GPa. By contrast, it is known that strongly hydrogen bonded crystals such as those comprising amino acids display Young's moduli greater than 25 GPa.40
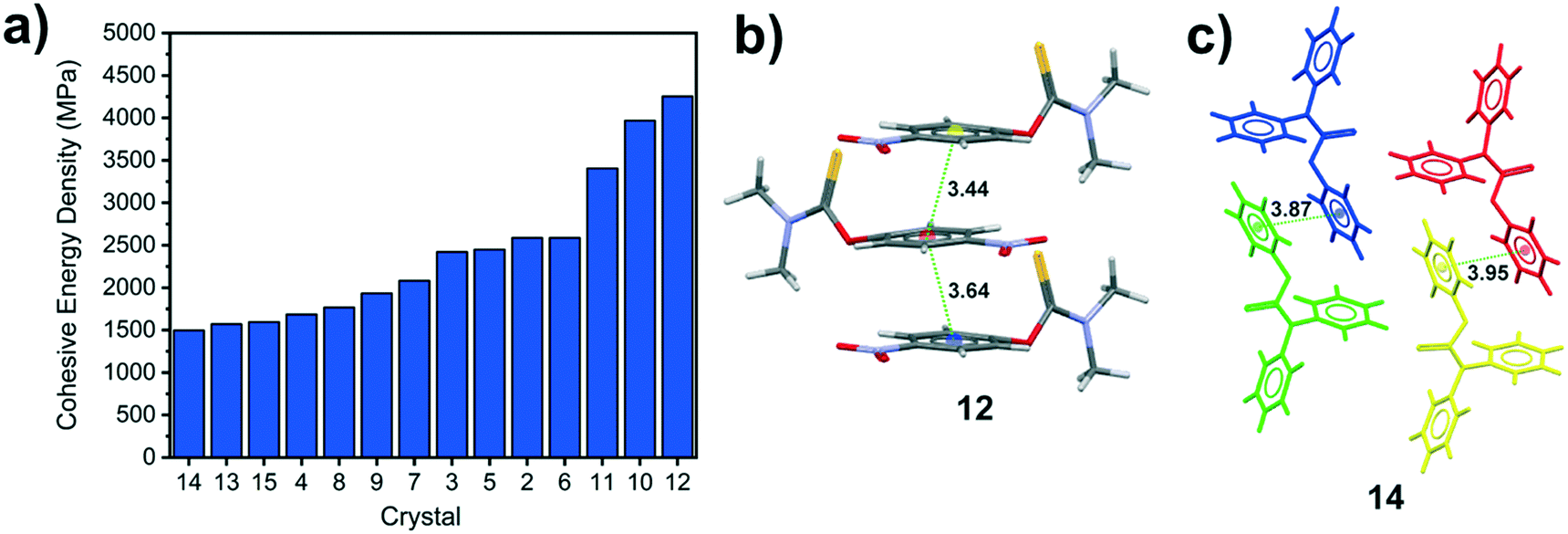 | ||
| Fig. 8 a) Calculated cohesive energy density for each crystal structure. Illustration of the efficient π⋯ π stacking interactions in the most stable b) crystal 12 compared to the stacking in c) crystal 14 where only one of the three aryl rings on each symmetry independent molecule engages in π⋯ π stacking interactions. Centroid–centroid distances are shown in cyan dotted lines with the distances reported in units of Å. | ||
Despite our efforts, we acknowledge that polymorphism might exist in these O-thiocarbamate systems. However, polymorphism was not observed experimentally from crystals that were grown from our limited selection of solvents that were able to afford diffraction quality crystals (please see the ESI†). The study of polymorphism in these systems is beyond the scope of this work but we cannot rule out that alternative polymorphic structures can exist for compounds 2–15.
Finally, Hirshfeld surface analysis (Fig. S33–S52†) was performed on all 14 crystal structures, as well as selected O-thiocarbamate compounds namely ERIWEH,41GIQDES,42JELTUQ,43JELVAY,43VIVVUU,44 and ZAHHUO,45 using CrystalExplorer.46 For most of the compounds analysed, the largest percentage contribution to the interatomic contacts of the Hirshfeld surfaces was mainly C–H/H–C (∼20–30%), H–H/H–H (∼15–65%) and H–S/S–H (10–20%) short-range contacts. Representative noteworthy close contacts are highlighted in Fig. 7, specifically for 14, 15, and JELTUQ. For instance, in compound 14, H⋯H interactions appear as the largest regions of the fingerprint plots (51.7%) with a high concentration at dexternal (de) = dinternal (di) ∼ 1.4–1.6 Å.
Of particular interest, are the red coloured regions on the Hirshfeld surface, which indicate short-range contacts (highlighted by red arrows in Fig. 9). These short-range interactions primarily correspond to C–H/H–C and H–H/H–H short contacts and can be attributed to intermolecular type III CH⋯π and van der Waals interactions. In some cases, such as 14 and 15, these regions are observed near the S atom, which correspond to two sharp spikes on the fingerprint plot (de + di ∼ 2.0 Å) due to weak CH⋯S interactions from the thiocarbamate–aryl stacking (in 15) or close proximity of the S atom with adjacent aryl CH or NCH3 moieties (in 14). Close contacts were also observed in 15 (de + di ∼ 1.9 Å), which are consistent with previously discussed aryl type III stacking from the naphthyl and phenyl moieties.
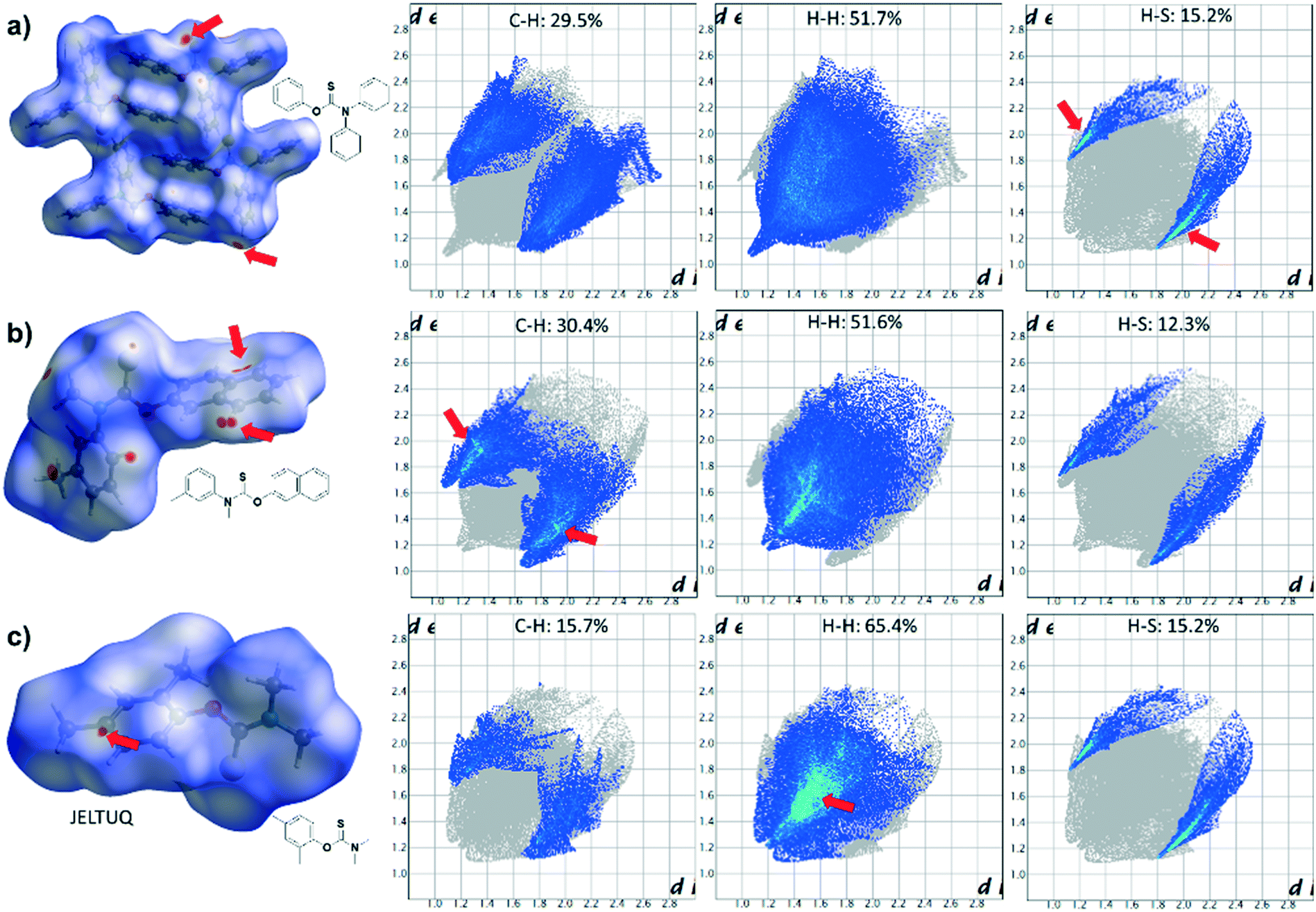 | ||
| Fig. 9 Hirshfeld surfaces mapped with dnorm (left) for (a) 14, (b) 15, and (c) JELTUQ, with their respective fingerprint plots showing the three largest percentage contributions of atoms (blue areas) within specific interacting pairs, namely C–H/H–C (left), H–H/H–H (middle), and H–S/S–H (right). | ||
In compounds with several CH3 groups on the phenyl and thiocarbamate moieties, such as JELTUQ, H⋯H interactions also contribute to the most significant percentage (65.4%) of the surface area with the highest concentration at de = di ∼ 1.4 Å. This is due to slipped-stacked type I and II packing motifs. For compounds containing Cl or F substituents (i.e., 2, 3, 6, 8, 10, and GIQDES) or strong HB acceptor moieties (i.e., 5, 9, 11, 12, ERIWEH, and VIVVUU), other types of interactions dominate their Hirshfeld surfaces.
Conclusions
In summary, we have presented the crystal structures and computed properties of a series of N,N-disubstituted-O-thiocarbamates that do not contain labile protic functional groups and have analysed how these molecules self-assemble in the solid-state in the absence of functional groups capable of engaging in directional hydrogen bonding interactions. We report for the first time the crystal structure of the anti-fungal tolnaftate drug. By performing a systematic joint experimental and theoretical study, we have identified four possible stacking modes that the molecules can engage in to form stable crystal structures. We have observed similar stacking patterns in the crystal structures of related molecules, particularly in cases where substituents are of comparable sizes, leading to isostructural crystals. The computed cohesive energy densities and mechanical properties of the crystals do not differ significantly amongst members of the series due to the weak and non-directional intermolecular forces in all the crystal structures. Despite the lack of formation of robust supramolecular building blocks that are typically found in hydrogen-bonded systems,47 we remain hopeful that our work in this system would inspire our fellow solid-state colleagues to look at similar biologically-active molecules that also do not contain typical HB motifs. Through understanding the self-assembly of such molecules, we hope to leverage the knowledge gained from this work as a platform, in the future, to create other O-thiocarbamate-based pharmaceutical solids such as cocrystals and salts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. G. would like to thank A*STAR AME IRG (A1783c0003) and a NTU start-up grant (M4080552) for financial support. H. S. S. is grateful for the Singapore Ministry of Education Academic Research Fund Tier 1 grants RG 111/18 and RT 05/19. H. S. S. also acknowledges that this project is supported by A*STAR under the AME IRG grants A1783c0003, A1783c0002, and A1783c0007. D. T. would like to thank A*STAR for a postdoctoral research fellowship. S. M. would like to acknowledge Khalifa University for financial support under the CIRA program (Project Code: CIRA-2018-068). The theoretical calculations were performed using the high-performance computing clusters of Khalifa University and the authors would like to acknowledge the support of the research computing department.Notes and references
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS
.
- B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS
- A. Nangia, J. Chem. Sci., 2010, 122, 295–310 CrossRef CAS
- C. B. Aakeröy, N. R. Champness and C. Janiak, CrystEngComm, 2010, 12, 22–43 RSC
- K. Biradha, C.-Y. Su and J. J. Vittal, Cryst. Growth Des., 2011, 11, 875–886 CrossRef CAS
- C. B. Aakeröy, T. K. Wijethunga and J. Desper, Chem. – Eur. J., 2015, 21, 11029–11037 CrossRef
- O. Bolton and A. J. Matzger, Angew. Chem., Int. Ed., 2011, 50, 8960–8963 CrossRef CAS
- W. Yang, A. Greenaway, X. Lin, R. Matsuda, A. J. Blake, C. Wilson, W. Lewis, P. Hubberstey, S. Kitagawa, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2010, 132, 14457–14469 CrossRef CAS
- T. Adachi and M. D. Ward, Acc. Chem. Res., 2016, 49, 2669–2679 CrossRef CAS
- S. Cherukuvada and T. N. G. Row, Cryst. Growth Des., 2014, 14, 4187–4198 CrossRef CAS
- F. Topić and K. Rissanen, J. Am. Chem. Soc., 2016, 138, 6610–6616 CrossRef
- D. Braga, L. Maini, F. Grepioni, A. De Cian, O. Félix, J. Fischer and M. W. Hosseini, New J. Chem., 2000, 24, 547–553 RSC
- M. K. Corpinot, S. A. Stratford, M. Arhangelskis, J. Anka-Lufford, I. Halasz, N. Judaš, W. Jones and D.-K. Bučar, CrystEngComm, 2016, 18, 5434–5439 RSC
- S. Mohamed, A. A. Alwan, T. Friščić, A. J. Morris and M. Arhangelskis, Faraday Discuss., 2018, 211, 401–424 RSC
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 31, 2311–2321 CrossRef
-
J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons, Ltd., United Kingdom, 2nd edn, 2009 Search PubMed
- O. V. Shishkin, R. I. Zubatyuk, S. V. Shishkina, V. V. Dyakonenkoa and V. V. Medviediev, Phys. Chem. Chem. Phys., 2014, 16, 6773–6786 RSC
- N. Blagden, M. de Matas, P. T. Gavan and P. York, Adv. Drug Delivery Rev., 2007, 59, 617–630 CrossRef CAS
- S. Karki, T. Friščić, L. Fábián, P. R. Laity, G. M. Day and W. Jones, Adv. Mater., 2009, 21, 3905–3909 CrossRef CAS
- D. J. Good and N. Rodríguez-Hornedo, Cryst. Growth Des., 2009, 9, 2252–2264 CrossRef CAS
- A. N. Sokolov, T. Friščić and L. R. MacGillivray, J. Am. Chem. Soc., 2006, 128, 2806–2807 CrossRef CAS
- H. Wang, G. Gurau, J. Shamshina, O. A. Cojocaru, J. Janikowski, D. R. MacFarlane, J. H. Davis Jr. and R. D. Rogers, Chem. Sci., 2014, 5, 3449–3456 RSC
- D. P. Ericson, Z. P. Zurfluh-Cunningham, R. H. Groeneman, E. Elacqua, E. W. Reinheimer, B. C. Noll and L. R. MacGillivray, Cryst. Growth Des., 2015, 15, 5744–5748 CrossRef CAS
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS
- S. J. Dalgarno, P. K. Thallapally, L. J. Barbour and J. L. Atwood, Chem. Soc. Rev., 2007, 36, 236–245 RSC
- K. J. Ardila-Fierro, V. André, D. Tan, M. T. Duarte, R. W. Lancaster, P. G. Karamertzanis and T. Friščić, Cryst. Growth Des., 2015, 15, 1492–1501 CrossRef CAS
- N. S. Ryder, I. Frank and M.-C. Dupont, Antimicrob. Agents Chemother., 1986, 29, 858–860 CrossRef CAS
- A. Bianchi, G. Monti and I. de Carner, Antimicrob. Agents Chemother., 1977, 12, 429–430 CrossRef CAS
- C. Faiman, R. J. Ryan and H. J. Eichel, Endocrinology, 1967, 81, 88–92 CrossRef CAS
- Derived from reported structures in the Cambridge Structural Database (CSD), CCDC SEARCH date 10 June 2020 .
- F. S. Kuan, F. Mohr, P. P. Tadbuppa and E. R. T. Tiekink, CrystEngComm, 2007, 9, 574–581 RSC
- N. H. Slater, B. R. Buckley, M. R. J. Elsegood, S. J. Teat and M. C. Kimber, Cryst. Growth Des., 2016, 16, 3846–3852 CrossRef CAS
- R. Custelcean, Chem. Commun., 2008, 295–307 RSC
- A. J. Perkowski, C. L. Cruz and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 50, 15684–15687 CrossRef
- T. A. Ablott, M. Turzer, S. G. Telfer and C. Richardson, Cryst. Growth Des., 2016, 16, 7067–7073 CrossRef CAS
- A. I. Kitaigorodskii, Acta Crystallogr., 1965, 18, 585–590 CrossRef CAS
- G. R. Desiraju and J. A. R. P. Sarma, J. Chem. Sci., 1986, 96, 599–605 CrossRef CAS
- N. K. Nath and A. Nangia, Cryst. Growth Des., 2012, 12, 5411–5425 CrossRef CAS
- M. Yamada, CCDC 1422039, Experimental Crystal Structure Determination, 2017, DOI:10.5517/ccdc.csd.cc1jqr77.
- I. Azuri, E. Meirzadeh, D. Ehre, S. R. Cohen, A. M. Rappe, M. Lahav, I. Lubomirsky and L. Kronik, Angew. Chem., Int. Ed., 2015, 54, 13566–13570 CrossRef CAS
- C. M. L. Vande Velde, H. J. Geise and F. Blockhuys, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, o199–o200 CrossRef CAS
- K. Eriksen, A. Ulfkjær, T. I. Sølling and M. Pittelkow, J. Org. Chem., 2018, 83, 10786–10797 CrossRef CAS
- A. Flores-Figueroa, V. Arista-M., D. Talancón-Sánchez and I. Castillo, J. Braz. Chem. Soc., 2005, 16, 397–403 CrossRef CAS
- D. Shi, S. Chen, B. Dong, Y. Zhang, C. Sheng, T. D. James and Y. Guo, Chem. Sci., 2019, 10, 3715–3722 RSC
- R. Zhang, L. Xu, H. Chen, Z. Qin, Y. Zhao and Z. Ni, Chem. Res. Chin. Univ., 2015, 31, 224–227 CrossRef CAS
-
M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, University of Western Australia, 2017, https://hirshfeldsurface.net Search PubMed
- D. Tan, Z. X. Ng, Y. Sim, R. Ganguly and F. García, CrystEngComm, 2018, 20, 5998–6004 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1831294–1831307. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce01336f |
| This journal is © The Royal Society of Chemistry 2020 |