
Halogen bonding between entirely negative fluorine atoms? Evidence from the crystal packing of some gold(I) and gold(III) complexes with extensively fluorinated m-terphenyl ligands and triphenylphosphane†‡
Alexandru
Sava
 ,
Krisztina T.
Kegyes
,
Bianca T.
Popuş
,
Bernadette C.
Dan
,
Cristian
Silvestru
and
Ciprian I.
Raţ
*
,
Krisztina T.
Kegyes
,
Bianca T.
Popuş
,
Bernadette C.
Dan
,
Cristian
Silvestru
and
Ciprian I.
Raţ
*
Supramolecular Organic and Organometallic Chemistry Centre, Chemistry Department, Faculty of Chemistry and Chemical Engineering, Babeş-Bolyai University, Cluj-Napoca RO-400028, Romania. E-mail: ciprian.rat@ubbcluj.ro; Fax: +40 (264) 590 818; Tel: +40 (264) 593 833
First published on 16th November 2020
Abstract
Despite lacking positive σ-holes, fluorine atoms of perfluorophenyl rings can engage in solid state fluorine–fluorine interactions, analogous to halogen bonding, in the absence of metal surface constraints or cryogenic temperatures. These interactions compete with other intermolecular interactions involving phenyl and perfluorophenyl rings for the supramolecular assembly of some gold(I) and gold(III) complexes.
Halogen bonding1 was considered for many years as the net attraction, primarily electrostatic,2 “between an electrophilic region on a halogen atom in a molecular entity and a nucleophilic region in another, or the same, molecular entity”.3 Although investigations on the nature of halogen bonding showed that it is largely dependent on both electrostatic and dispersion components,4 following the development of the σ-hole concept,5 the attraction was attributed to electrostatics. Fluorine, being the most electronegative element, seldom develops a positive σ-hole, thus leaving only a few instances when it was recognized as either halogen bond donor or acceptor,6 some even questioning fluorine's ability to participate in halogen bonding.7
Solid state non-bonded F⋯F interactions, although considered for a long time the result of crystal packing,8 have been periodically reported and rediscovered.9 Some reports included notable examples of F⋯F contacts occurring without further support,9d,f suggesting that they could be an influencing factor in the packing motif. Following a more recent report on the self-assembly of fully fluoro-substituted phenyleneethynylene molecules, deposited on a Ag(111) surface at low temperature,10 F⋯F interactions started receiving considerably more attention.11 These molecules were found to self-assemble, attracted by dispersion forces between fluorine atoms which were surrounded by an entirely negative, anisotropically distributed, electrostatic potential. Although X⋯X (X = Cl, Br and I) bonding having similar directionality had previously been reported, authors concluded that the interaction was only similar to halogen bonding, because the measured F⋯F distance was “at least 300 pm, slightly larger the twice the vdW radius of a fluorine atom (i.e., 294 pm)”.
Further complementary evidence came from real-space imaging of the intermolecular bonding structures in the two dimensional self-assembly of C6F6 and C6Br6 molecules, adsorbed on a Ag(110) surface at 600 mK.11c Once again, the images showed a similar directionality in the bonding patterns, suggesting a “common bonding mechanism”, despite the fact that fluorine atoms were “not expected to develop a (positive) σ-hole”. This counter-intuitive attraction between atoms lacking positive electrostatic potential12 was further explored and confirmed computationally.11a,e
Placing such molecules on metal surfaces, however, influences their degrees of freedom and disfavours the formation of attractive C–F⋯πF (πF = πperfluoroaryl) interactions,13 which usually dominate the crystal packing of perfluorinated molecules. On the metal surface, the molecules approached side by side and the “unduly underestimated” dispersion forces14 took charge of the assembly. In the present work, we report solid state evidence for the F⋯F interactions between entirely negative fluorine atoms of perfluorophenyl rings, in the absence of metal surface constraints or cryogenic temperatures. Directional F⋯F bonding, at distances below ΣrvdW(F, F), connects supramolecular entities in the crystal, without requiring further support from other significant intermolecular interactions, thus confirming their attractive nature. These interactions compete with, or even overshadow, other potentially stronger non-covalent interactions (NCIs).
We have focused recently on the synthesis of fluorinated ligands with m-terphenyl backbone. Expanding from previously reported 2,4,6-(C6F5)3C6H2Br (1c),15 we prepared two other proligands: 2,6-(C6F5)2-4-MeC6H2Br (1a) and 2,6-(C6F5)2C6F3Br (1b)16 in Ullmann-type cross coupling reactions from the corresponding aryl halides III and IV (Scheme S1 in ESI‡). These proligands were used to synthesize gold(I) complexes with triphenylphosphane (2a–c), which were subsequently oxidised with PhICl2 to their gold(III) analogues (3a–c) (Scheme 1).
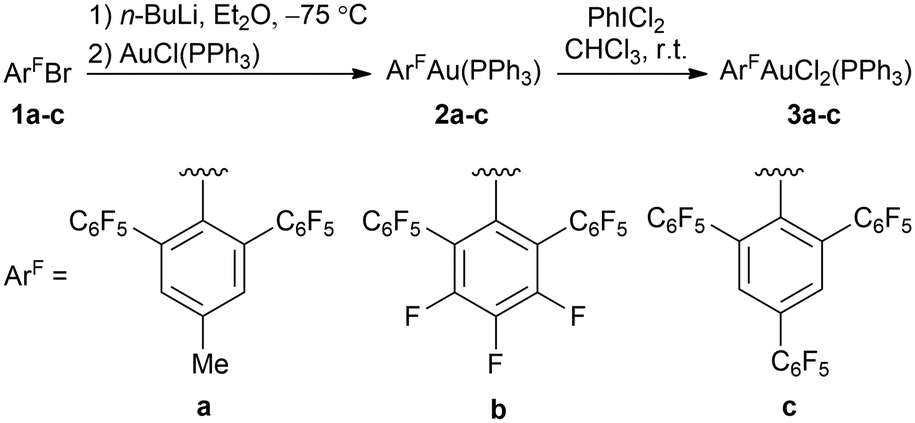 | ||
| Scheme 1 Synthesis of gold complexes 2a–c and 3a–c. | ||
Solution NMR spectra show unexpected resonances only for gold(III) complexes 3a–c. The cis addition of chlorine atoms causes steric crowding, which leads to restricted rotation around Au–P and Au–C bonds, as well as for the flanking C6F5, around the corresponding C–C bonds within the organic ligand attached to the metal atom. A really remarkable feature in the 13C{1H} NMR spectra of 3a–c is the presence of a through-space spin–spin 13C–19F coupling,17 at eight covalent-bond separation between atoms (discussion and Fig. S1 in ESI‡).
In the solid state, gold(I) complexes 2a–c display linear coordination geometry while gold(III) complexes 3a–c are square-planar with chlorine atoms cis to each other (for molecular parameters, see Table S2 in ESI‡). The molecular structure of 3b·CHCl3 is illustrated in Fig. 1 (for 2a–c, 3a and 3c·CHCl3, see Fig. S4–S8 in ESI‡). Around gold atom, C(19)–Au(1)–P(1) angle increases to 97°, while Cl(1)–Au(1)–Cl(2) drops below 90°. Around phosphorus atom, Au(1)–P(1)–C(1) angle increases up to 120°. A comparison between the structural parameters of 3a–c with those of the related cis-[(2,4,6-C6F3H2)AuCl2(PPh3)] (CCDC-BOGKUF),18cis-[(C6F5)AuCl2(PPh3)] (CCDC-TIFSEI),19 and cis-[(C6F5)AuCl2(PtBu)3] (CCDC-GILKOD)20 (Table S2 in ESI‡) reveals the extent of steric clash between the phenyl ring stuck above the fluorinated ligand and the flanking C6F5 groups. This proximity leads to the aforementioned through-space spin–spin coupling, between C(2)/C(6) and F(1)/F(6), in all gold(III) complexes.
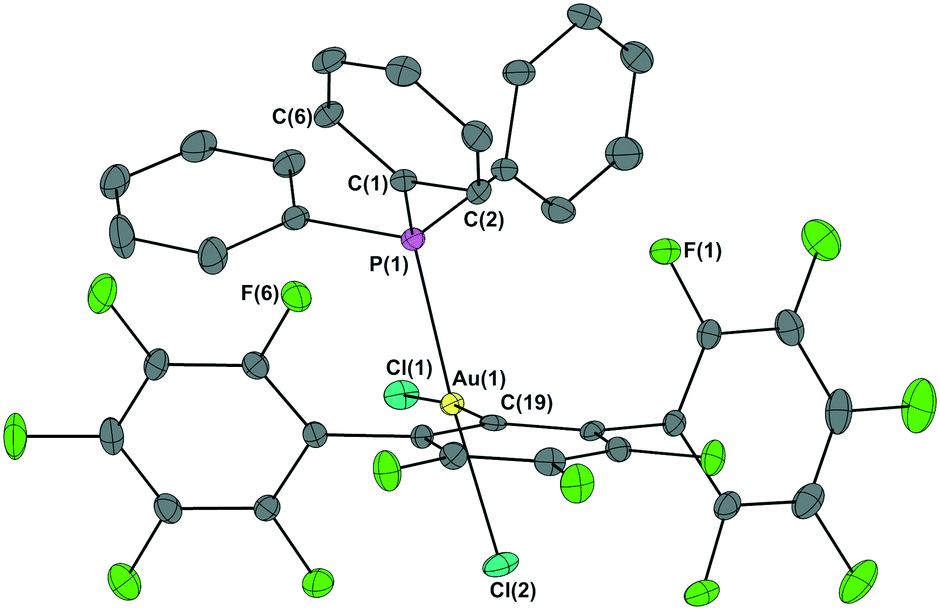 | ||
| Fig. 1 Thermal ellipsoid representation of the solid state structure of cis-[{2,6-(C6F5)2C6F3}AuCl2(PPh3)] (3b·CHCl3). Ellipsoids are drawn at 30% probability level. Hydrogen atoms and the solvent molecule were omitted for clarity. | ||
We sought to identify the most significant intermolecular interactions responsible for the supramolecular assembly of all gold complexes. A first, surprising, observation was that all the assemblies are dominated by fluorine-centred C–F⋯πF, C–F⋯H–C and F⋯F interactions,21 rather than π⋯π stacking,22 C–H⋯π (ref. 23) or C–Cl⋯π (ref. 24) interactions (π = πF or πH; πH = C6H5). While C–F⋯πF are known to be attractive, C–F⋯πH interactions are repulsive.13 Lone-pair⋯π, including C–F⋯πF and C–Cl⋯π, interactions are considered significant “if the distance separating the electron-rich atom from one atom of the six-membered ring is in the range of the sum of their van der Waals radii”.25 Regarding C–F⋯H–C interactions, in a comprehensive study,26 the peak distribution for F⋯H distances was found at 2.40 Å, with more than 98% distances shorter than 2.56 Å. Thus, we focused on interatomic distances that were less than ΣrvdW, as it is recommended for halogen bonding as well3 (rF = 1.47 Å, rC = 1.70 Å, rCl = 1.75 Å,27 and rH = 1.10 Å (ref. 28)). Intermolecular F⋯F interactions (ΣrvdW(F, F) = 2.94 Å), i.e. not complementary to the stronger C–F⋯πF interactions,29 of both type I and II,30 are listed in Table 1. Intermolecular C–X⋯H–C, Au–Cl⋯H–C (ΣrvdW(H, F) = 2.57 Å, ΣrvdW(H, Cl) = 2.85 Å, Table S3‡) and C–F⋯πF (ΣrvdW(C, F) = 3.17 Å, Table S4‡) interactions in all complexes are listed in ESI.‡ A few observed C–H⋯π interactions (Table S5‡) and π⋯π stacking (Table S6‡) are also listed in ESI.‡ No C–Cl⋯π (ΣrvdW(C, Cl) = 3.45 Å) were observed at all.
| Entry | Compound | F⋯F (Å) | θ 1 (deg) | θ 2 (deg) | Type |
|---|---|---|---|---|---|
| 1 | 3a | 2.906(10) | 106.3(6) | 91.5(6) | I |
| 2 | 2b | 2.810(6) | 161.7(3) | 102.6(4) | II |
| 3 | 2.882(5) | 125.7(3) | 100.4(4) | I | |
| 4 | 2.904(5) | 95.7(3) | 123.7(4) | I | |
| 5 | 3b | 2.817(6) | 147.0(3) | 136.3(3) | I |
| 6 | 2.860(6) | 169.8(4) | 168.0(3) | I | |
| 7 | 2.900(5) | 124.6(3) | 174.2(3) | II | |
| 8 | 2c | 2.685(12) | 145.8(9) | 115.7(7) | II |
| 9 | 2.912(10) | 140.2(6) | 140.2(6) | I | |
| 10 | 2.927(9) | 116.2(5) | 159.2(6) | II | |
|
|||||
The supramolecular arrangement of molecules of 2a, 3a and 3c is driven by C–F⋯πF and C–F⋯H–C interactions with negligible contributions from C–H⋯π and F⋯F interactions. For molecules of 2b, we found significant F⋯F interactions. One F⋯F interaction (2.810 Å) is type II which, in the case of Cl⋯Cl is “unambiguously considered as attractive”.31 Nonetheless, this is a typical case when the contribution of F⋯F interactions to the construction of the 3D network cannot be assessed separately, as in the same direction with each F⋯F interaction, other NCIs provide support. Numerous C–F⋯πF, C–F⋯H–C and C–H⋯π interactions span the 3D network in all directions.
On the other hand, significant F⋯F interactions were also found in the crystal structure of 3b, but, in contrast with 2b, their contribution to the construction of the 3D network is very well defined. Layers of molecules built through C–F⋯πF, C–Cl⋯H–C and C–F⋯H–C interactions (Fig. S9 in ESI‡), are assembled by inter-layer F⋯F interactions (one at 2.817 Å and two coplanar at 2.860 Å and 2.900 Å, for each molecule; cf. ΣrvdW(F, F) 2.94 Å) (Fig. 2 – a axis, and Fig. S10 in ESI‡ – b axis). Apart from F⋯F interactions, between the layers there is only a type I, F⋯Cl (3.11 Å; ΣrvdW(F, Cl) 3.22 Å), hetero-halogen bonding interaction6c (Fig. S11 in ESI‡); the shortest F⋯H contact is at 2.64 Å (cf. ΣrvdW(F, H) 2.57 Å) and no C–F⋯πF interactions were observed at all. In other words, the 2D sheets are kept close to each other by extended, cooperative, fluorine-centred halogen bonding interactions, at distances below ΣrvdW(F, X) (X = F, Cl). At the centre of the unit cell (Fig. 2) there are synergic, type II, F⋯F interactions (2.900 Å), at parameters in agreement with computational calculations.11e A Cambridge Structural Database32 search confirms that this supramolecular synthon (dF⋯F < ΣrvdW(F, F) 2.94 Å; θ2 > 170°) could be found in the crystals of 136 compounds.
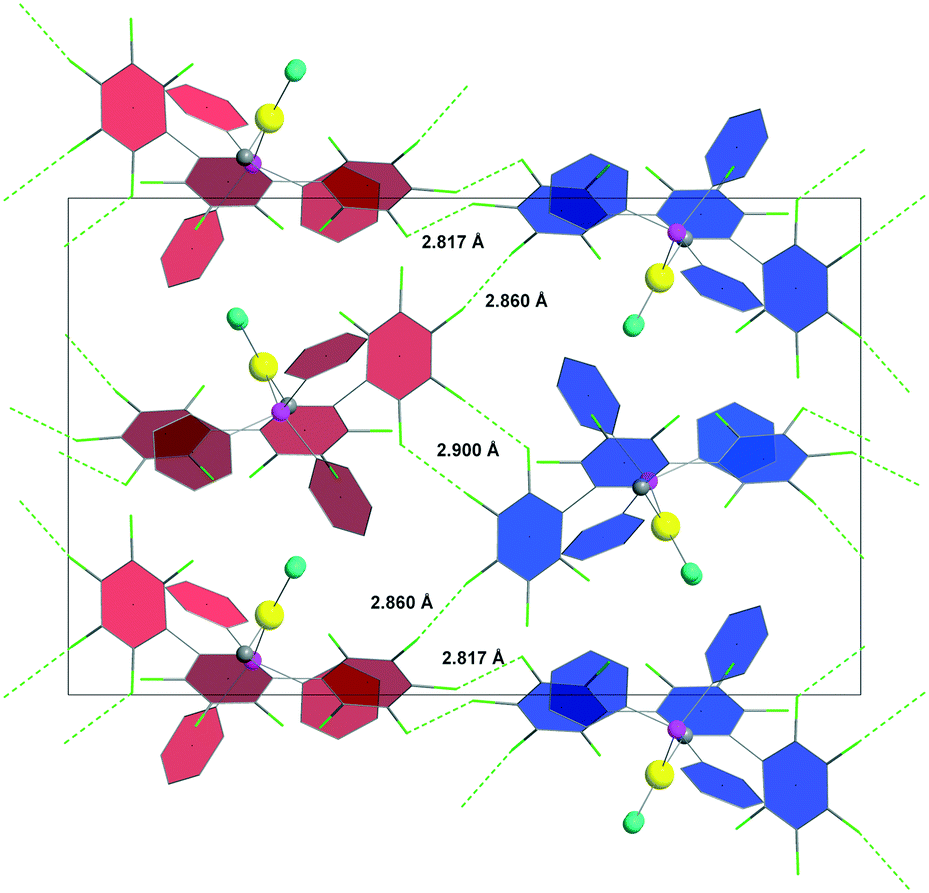 | ||
| Fig. 2 Two layers of molecules of 3b (red and blue phenyl rings for different layers) connected by cooperative F⋯F interactions (view along a axis). Intra-layer interactions i.e. C–F⋯πF, C–Cl⋯H–C and C–F⋯H–C, were omitted for clarity. Colour coding: Au = yellow, C = grey, Cl = turquoise, P = pink. | ||
Further evidence is brought by the solid-state structure of 2c. Besides usual C–F⋯πF or C–F⋯H–C interactions, there are also F⋯F intermolecular interactions (2.685 Å, 2.912 Å and 2.927 Å). Using the Crystal Explorer software package33 we were able to quantify the intermolecular contacts in the crystal structure of 2cvia the Hirshfeld surface analysis.34 From various parameters that can be extracted from the Hirshfeld surface analysis, the most relevant is the normalised contact distance dnorm, which takes into account the relative sizes of atoms, effectively highlighting the closest contacts.35
Following Hirshfeld analysis for molecules of 2c, the type II F⋯F interaction at 2.685 Å was found to be the shortest intermolecular contact (Fig. 3). This is impressive, because the most negative dnorm value is usually found for the strongest intermolecular interaction, as it is for example, O⋯H hydrogen bonding in paracetamol and oxalic acid.35 Another type II F⋯F contact having similar dnorm parameters was previously reported for molecules of 4,4′-bis(perfluorophenyl)-2,2′-bithiazole (PFBT).9i Moreover, in our case, the probability to find a F⋯F contact with the most negative dnorm value is significantly reduced as the ratio F/H is 15/17, compared to 10/2 in PFBT molecules, thus competing with potential C–F⋯H–C interactions. Remarkably, the crystal architecture of 2c is built from chains of molecules (Fig. S12, in ESI‡), connected solely by these type II F⋯F interactions at 2.685 Å (Fig. 4).
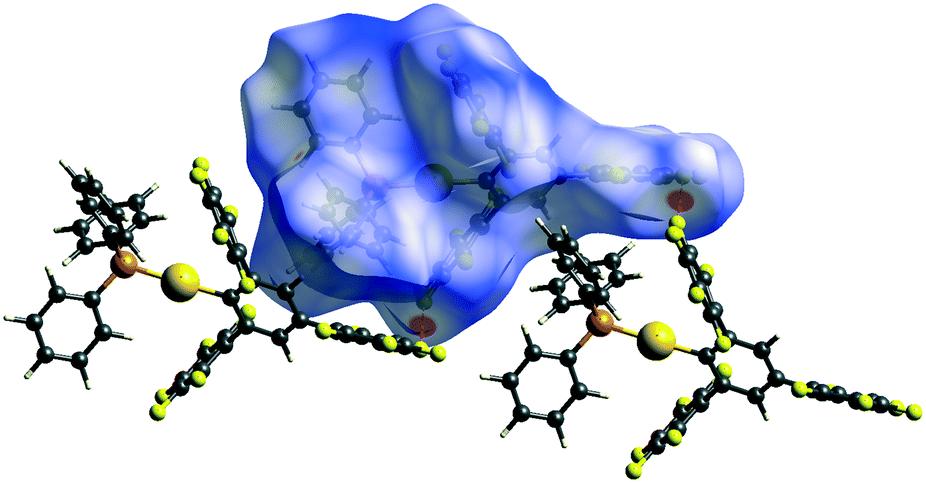 | ||
| Fig. 3 The dnorm mapped on Hirshfeld surfaces for molecules of 2c. The dnorm is displayed using red–white–blue colors, where red is for shorter contacts, i.e. negative dnorm values, white for contacts around the vdW separation, and blue for longer contacts, i.e. positive dnorm values. The most significant supramolecular interaction is the type II F⋯F interaction at 2.685 Å. | ||
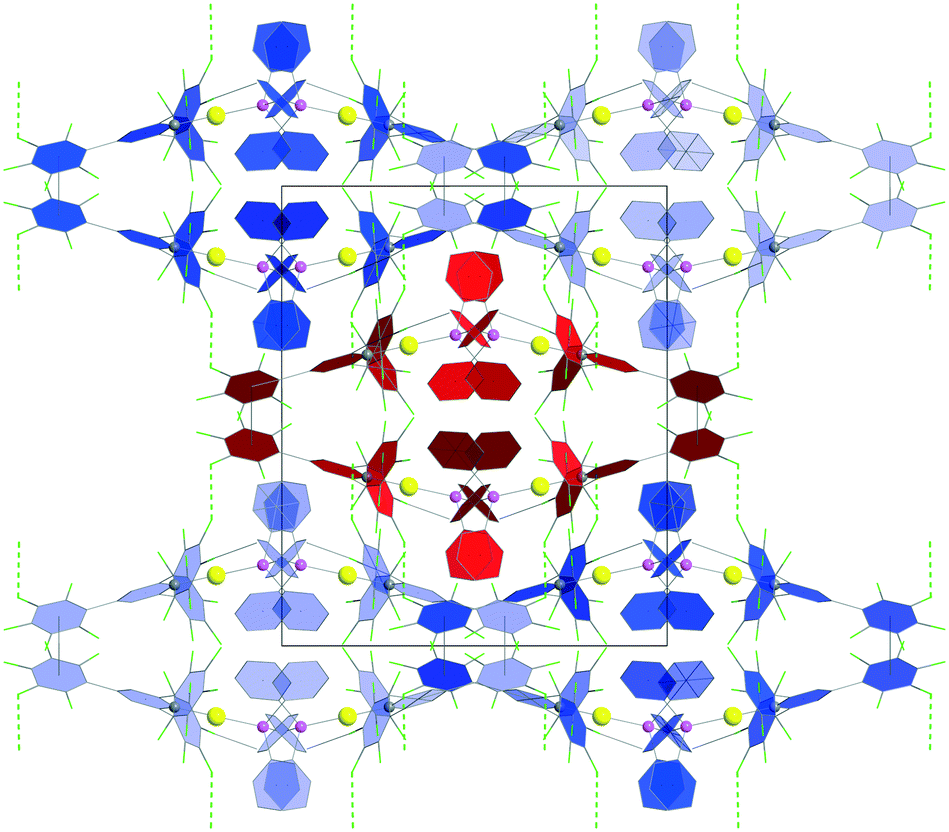 | ||
| Fig. 4 View along the supramolecular chains (c axis) of 2c. The chains are connected solely by type II F⋯F interactions at 2.685 Å. Colour coding: Au = yellow, C = grey, P = pink. | ||
Between the chains the closest C–F⋯H–C and C–F⋯C(πF) contacts are at 2.68 Å and at 3.31 Å, respectively.
DFT calculations of complexation energies were carried out on pairs of molecules generated using CrystalExplorer (see ESI,‡ page 64). In all the pairs that exhibit F⋯F contacts, the theoretical complexation energies indicate attractive interactions. Nevertheless, it should be noted that complexation energies give the overall interaction energy between molecules and not only the energy stemming from F⋯F contacts. Properties of bond critical point (BCP) of F⋯F interactions are in the range of those reported in the literature,36 and indicate a closed shell attractive character (see ESI,‡ Tables S8 and S9).
In conclusion, we have presented solid state evidence for considering F⋯F interactions as potential halogen bonds. Although fluorine atoms in common organic molecules lack positive σ-holes, which rather explains their poorer directionality,4c these unsupported interactions, mainly dispersive in nature, at distances below ΣrvdW, contribute to the solid-state arrangements, in some instances standing out from the bulk of all the other supramolecular interactions. As halogen bonding strongly depends not only on electrostatics, but also on dispersion and polarization,1b,4 the pieces of evidence we report herein complement previous observations on metal surfaces and are all prerequisites for recognition of halogen bonding phenomena3 for entirely negative fluorine atoms.
We thank the National University Research Council of Romania CNCS (research project no. PN-II-RU-TE-2014-4-0906) for financial support, Dr. Alexandra Pop, Dr. Albert Soran and Dr. Sergiu Shova for XRD determinations, and Clément Orione for recording the HOESY spectra. Theoretical calculations were performed on the high-performance computational facility MADECIP, POSCCE, COD SMIS 48801/1862 co-financed by the European Regional Development Fund of the European Union.
Conflicts of interest
There are no conflicts to declare.Notes and references
-
(a) P. Metrangolo and G. Resnati, Chem. – Eur. J., 2001, 7, 2511 CrossRef CAS
; (b) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478 CrossRef CAS
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748 RSC
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711 CAS
-
(a) K. E. Riley and P. Hobza, J. Chem. Theory Comput., 2008, 4, 232 CrossRef CAS
-
(a) T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291 CrossRef CAS
-
(a) P. Metrangolo, J. S. Murray, T. Pilati, P. Politzer, G. Resnati and G. Terraneo, CrystEngComm, 2011, 13, 6593 RSC
- K. Eskandari and M. Lesani, Chem. – Eur. J., 2015, 21, 4739 CrossRef CAS
- G. R. Desiraju and R. Parthasarathy, J. Am. Chem. Soc., 1989, 111, 8725 CrossRef CAS
-
(a) J. Kowalik, D. VanDerveer, C. Clower and L. M. Tolbert, Chem. Commun., 1999, 2007 RSC
- S. Kawai, A. Sadeghi, F. Xu, L. Peng, A. Orita, J. Otera, S. Goedecker and E. Meyer, ACS Nano, 2015, 9, 2574 CrossRef CAS
-
(a) P. R. Varadwaj, A. Varadwaj and B.-Y. Jin, Phys. Chem. Chem. Phys., 2015, 17, 31624 RSC
-
(a) J. Lekner, Proc. R. Soc. A, 2012, 468, 2829 CrossRef
-
(a) S. Kawahara, S. Tsuzuki and T. Uchimaru, J. Phys. Chem. A, 2004, 108, 6744 CrossRef CAS
- J. P. Wagner and P. R. Schreiner, Angew. Chem., Int. Ed., 2015, 54, 12274 CrossRef CAS
-
(a) M. Olaru, J. Beckmann and C. I. Raţ, Organometallics, 2014, 33, 3012 CrossRef CAS
- S. C. Cohen, A. J. Tomlinson, M. R. Wiles and A. G. Massey, J. Organomet. Chem., 1968, 11, 385 CrossRef CAS
-
(a) F. R. Jerome and K. L. Servis, J. Am. Chem. Soc., 1972, 94, 5896 CrossRef CAS
- M. Hofer, E. Gomez-Bengoa and C. Nevado, Organometallics, 2014, 33, 1328 CrossRef CAS
- M. Hofer and C. Nevado, Eur. J. Inorg. Chem., 2012, 1338 CrossRef CAS
- M. Hofer and C. Nevado, Tetrahedron, 2013, 69, 5751 CrossRef CAS
- K. Reichenbächer, H. I. Süss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22 RSC
-
(a) M. O. Sinnokrot, E. F. Valeev and C. D. Sherrill, J. Am. Chem. Soc., 2002, 124, 10887 CrossRef CAS
- M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873 RSC
- H. Sun, A. Horatscheck, V. Martos, M. Bartetzko, U. Uhrig, D. Lentz, P. Schmieder and M. Nazaré, Angew. Chem., Int. Ed., 2017, 56, 6454 CrossRef CAS
- T. J. Mooibroek, P. Gamez and J. Reedijk, CrystEngComm, 2008, 10, 1501 RSC
- E. D'Oria and J. J. Novoa, CrystEngComm, 2008, 10, 423 RSC
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS
- R. S. Rowland and R. Taylor, J. Phys. Chem., 1996, 100, 7384 CrossRef CAS
- R. M. Osuna, V. Hernández, J. T. López Navarrete, E. D'Oria and J. J. Novoa, Theor. Chem. Acc., 2011, 128, 541 Search PubMed
- N. Ramasubbu, R. Parthasarathy and P. Murray-Rust, J. Am. Chem. Soc., 1986, 108, 4308 CrossRef CAS
- T. T. T. Bui, S. Dahaoui, C. Lecomte, G. R. Desiraju and E. Espinoza, Angew. Chem., Int. Ed., 2009, 48, 3838 CrossRef CAS
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, University of Western Australia, Perth, Australia, 2017 Search PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19 RSC
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814 RSC
- G. V. Janjić, S. T. Jelić, N. P. Trišović, D. M. Popović, I. S. Đorđević and M. K. Milčić, Cryst. Growth Des., 2020, 20, 2943 CrossRef
Footnotes |
| † Dedicated to Professor Antonio Laguna as a recognition of his outstanding contributions to gold chemistry and his support for the development of this research field at Babeş-Bolyai University. |
| ‡ Electronic supplementary information (ESI) available: Synthetic details, NMR, MS and crystallographic data, including numbering scheme for NMR resonances assignments, tables of intermolecular interactions and figures representing the supramolecular architectures in the crystal and details of the theoretical study. CCDC 1985132 (2a), 1981844 (2b), 1985133 (2c), 1981845 (3a), 1981847 (3b·CHCl3), 1981846 (3c·CHCl3). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce00671h |
| This journal is © The Royal Society of Chemistry 2020 |