
Increasing the performance, trustworthiness and practical value of machine learning models: a case study predicting hydrogen bond network dimensionalities from molecular diagrams†‡
Andre P.
Frade
 a,
Patrick
McCabe
b and
Richard I.
Cooper
*a
a,
Patrick
McCabe
b and
Richard I.
Cooper
*a
aChemical Crystallography Laboratory, Department of Chemistry, University of Oxford, UK. E-mail: richard.cooper@chem.ox.ac.uk; Tel: +44 (0)1865 285000
bCambridge Crystallographic Data Centre, 12 Union Road, Cambridge, UK
First published on 12th March 2020
Abstract
The performance of a model is dependent on the quality and information content of the data used to build it. By applying machine learning approaches to a standard chemical dataset, we developed a 4-class classification algorithm that is able to predict the hydrogen bond network dimensionality that a molecule would adopt in its crystal form with an accuracy of 59% (in comparison to a 25% random threshold), exclusively from two and lower dimensional molecular descriptors. Although better than random, the performance level achieved by the model did not meet the standards for its reliable application. The practical value of our model was improved by wrapping the model around a confidence tool that increases model robustness, quantifies prediction trust, and allows one to operate a classifier virtually up to any accuracy level. Using this tool, the performance of the model could be improved up to 73% or 89% with the compromise that only 34% and 8% of the total set of test examples could be predicted. We anticipate that the ability to adjust the performance of reliable 2D based models to the requirements of its different applications may increase their practical value, making them suitable to tasks that range from initial virtual library filtering to profile specific compound identification.
Introduction
Cheminformatics models promise to deliver detailed information about compounds in a fraction of the time and resources required by traditional methods that often involve compound synthesis and experimental property determination.1 However, some would argue that the majority of published models do not meet the requirements for a reliable practical use.2,3 The core of poor performance often stems from limitations in the data used for model production, whether it relates to its poor quality3,4 or lack of information content that is relevant to the property being modelled.5Most property prediction models are produced from feature vector representations.6 These consist of arrays of numbers representing chemical structure descriptors, such as molecular weight or number of hydrogen bond donors, which together build a molecule's profile. Different types of descriptors are available. Two and lower dimensional descriptors are those that can be rapidly derived from molecular formulas and diagrams at low computational cost. Despite their deterministic unambiguous computation, these are often limited in their information content, usually lacking any 3D-spatial arrangement information of the atoms.6 Given that molecules can co-exist in multiple conformations, these descriptors may be insufficient to fully describe a given property,7 especially those to which conformational flexibility is highly relevant.8,9 On the other hand, three and higher dimensional descriptors are able to capture the three-dimensional conformation of molecules and their interaction with the environment.6,10 Despite their high information content, these descriptors rely on the atomic coordinates of compounds, whose prediction is computationally expensive and cannot be guaranteed to correspond to the relevant conformation,1,3,11,12 which can considerably increase the runtime of the algorithm without adding any useful contribution,7 or even decreasing model performance.13 The deterministic character and potential information content of descriptors are key factors to consider during descriptor selection, as it will have implications on property description, but also on model performance, robustness and stability.9,14
The accuracies of models exclusively built from two and lower dimension descriptors tend to be lower, yet we believe that those performing reasonably better than random have an underestimated potential that is often left unexplored. Thus, we suggest the development of strategies that allow the exploitation of the prediction mechanism to provide valuable guidance on how to improve the model performance and its practical value.15,16
Hydrogen bond network dimensionality (HBND) describes how hydrogen-bond intermolecular interactions extend in a three-dimensional structure. The network expansion is guided by the set of available hydrogen bonding groups in a molecule and their allowed interactions.17 The resulting dimensionality is thought to be a major cause of anisotropic interactions in crystal structures due to its directional nature.18,19 Although its impact is not well characterised, dimensionalities often act as valuable complementary information to the study of properties that are directly influenced by slip plane arrangements in crystals, such as crystal stability, mechanical behaviour and tabletability performance.17,20,21
Bryant et al.17 recently described an automated method to assign the dimensionality of a hydrogen bond network from solved crystal structures, and have demonstrated its effectiveness on multiple drug systems by comparison with tabletability data. However, the reliance on solved crystal structures as input limits the large scale implementation of the tool. Crystals of compounds of interest are rarely available, obtaining them is resource and time consuming, and crystal structure predictions are computationally expensive and still not reliable enough for such application.3,11
Machine learning predictive models have been widely adopted as a good alternative to experimental property determination, and 2D based quantitative structural property relationship models (QSPRs) become particularly useful in the HBND context. The hydrogen bond network dimensionality problem can be formulated as a four-class classification task, and the four possible network dimensionality outcomes are schematically represented in Fig. 1. In this work we present the possibility of hydrogen bond network dimensionality prediction to any region of chemical space, such that the screening of large virtual libraries becomes feasible and reliable. We further develop and test a confidence measure that adds robustness to classification algorithms and quantifies the trust of each output prediction. The tool also enables one to adjust the compromise between accuracy level and prediction output accessibility that best suits the requirements of the context under which the model is used. This approach may enable additional 2D-based model applications, such as robust single molecule property prediction or production of structure–property relationship insights.
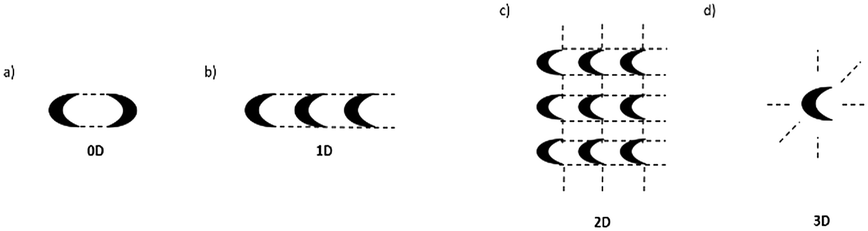 | ||
| Fig. 1 Hydrogen bonding possible interactions and resulting network dimensionality. a) Zero dimensional, 0D, comprised of enclosed motifs such as rings. b) One dimensional, 1D, chains. c) Two dimensional, 2D, sheets. d) Three dimensional, 3D, when the network expands in all possible directions. | ||
Results and discussion
The dataset
Only organic crystals with a single chemical component were considered, as described in the Methods section. Data collection, cleansing and scaling resulted in a final dataset of 64![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 084 dimensionality labelled examples – 22767 with 0D networks, 30943 with 1D networks, 7123 with 2D networks, and 3251 with 3D networks – described by a set of 113 numerical descriptors. From this, a class-balanced dataset was produced by random under-sampling of over-represented classes, giving a total of 13004 scaled examples evenly distributed across the 4 classes. The two dimensional visualization of the balanced dataset by t-SNE (Fig. 2) shows that data points are evenly distributed, which visually does not suggest that our data provides a good separation between classes.
084 dimensionality labelled examples – 22767 with 0D networks, 30943 with 1D networks, 7123 with 2D networks, and 3251 with 3D networks – described by a set of 113 numerical descriptors. From this, a class-balanced dataset was produced by random under-sampling of over-represented classes, giving a total of 13004 scaled examples evenly distributed across the 4 classes. The two dimensional visualization of the balanced dataset by t-SNE (Fig. 2) shows that data points are evenly distributed, which visually does not suggest that our data provides a good separation between classes.
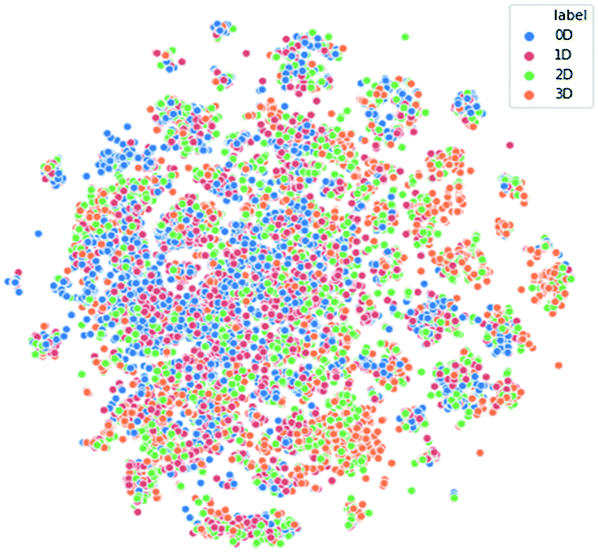 | ||
| Fig. 2 Visualization of the hydrogen bond network dimensionality dataset by t-SNE. Each data point is an entry of the balanced dataset that has been compressed from an original 113 dimensional space to two dimensions, and it is coloured according to the class it belongs to. | ||
Better than random studies
The balanced dataset was used to train a selection of statistical models (ESI‡ B). Accuracy predictions of random data were compared against true test data to determine the random performance threshold and check whether models perform better than that.As expected, the random performance threshold was found to correspond to 25% accuracy. All models performed considerably better than random, providing evidence that the data is indeed informative of the property (ESI‡ B).
Model optimisation and selection
In order to find the statistical method and optimal hyper-parameters that best suit the classification task, all methods were reconsidered and subject to a hyper-parameter grid search optimization. Mean cross validation accuracies were used to compare model performances, to reduce any accuracy bias towards a particular dataset split.All optimized models achieved similar results (ESI‡ B). The multiclass implementation of the SVM RBF (radial basis function kernel) slightly outperformed the others, attaining a total accuracy of 59% on the test set. The corresponding confusion matrix can be seen in Fig. 3, left. The classifier is able to detect each class with an accuracy considerably higher than random (random accuracy value of 0.25). The model was further tested on the 51080 examples discarded during class size balancing, where the accuracy per class remained effectively unchanged, demonstrating the generalization capability of the model. Ultimately, these findings suggest that hydrogen bond network dimensionality can be approximately estimated from two dimensional molecular descriptors. We also notice that misclassified examples tend to be assigned to adjacent classes, suggesting that the definition of network dimensionalities is a continuum, and so there isn't a well-defined boundary between adjacent classes.
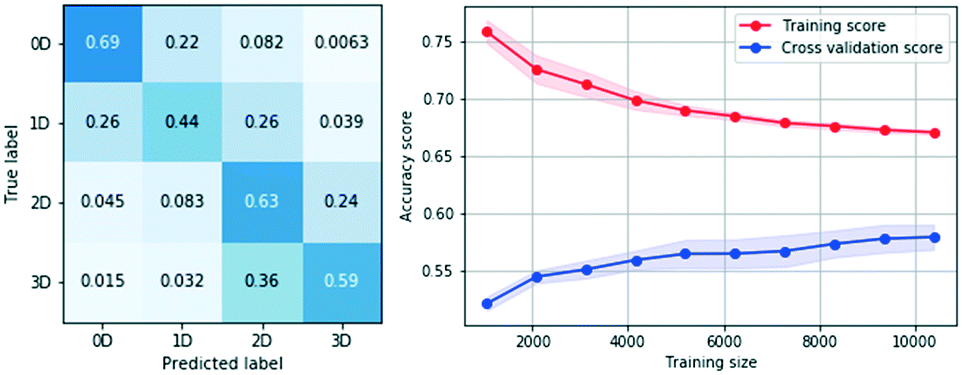 | ||
| Fig. 3 Confusion matrix and learning curve results for SVM RBF models trained on two dimensional descriptors. The standard deviations of the learning curve values are indicated by the shaded areas. | ||
The learning curve (Fig. 3, right) shows that the model tends to generalise well to unseen examples, suggesting that no overfitting has occurred during the training stage. The lack of convergence between training and cross validation score lines shows high variance and suggests that the model performance could be improved. Learning curves built from accuracy scores also provide upper bounds for how good a model can get using the set of descriptors considered. The upper bound corresponds to the accuracy at which both line scores would theoretically converge, which we estimate from the learning curve to be between 60% and 65%. As expected, these findings confirm the limitations of predicting three dimensional properties like hydrogen bond network dimensionality exclusively from two and lower dimensional molecular descriptors. Such datasets may be incomplete in scenarios where, for example, a single compound defined by a unique set of two and lower dimensional features may have the ability of adopting different packing arrangements (polymorphs) which may lead to different network dimensionalities in their crystal form.17
Confidence thresholds
We introduce the notion of confident thresholds and confident guesses, to develop a confidence restriction measure that increases the trust of predictions of any classification model. In these settings, each prediction output consists of the array of probabilities of an example belonging to each class. A confident threshold is the minimal gap that must exist between the two highest probabilities in this array, and a confident guess occurs when this gap is satisfied. If so, the example is assigned to the most probable class. The test examples that the estimator is not able to predict with confidence are not classified. These are stored and can be passed onto another model, or to the same model subject to a lower confidence threshold. The confidence restriction measure was tested on the hydrogen bond network dimensionality dataset and SVM RBF predictive model.First we investigated the benefits of considering confidence thresholds. To determine how many predictions were facing a small probability difference between their two most probable classes, the test set was first evaluated by the model with no confidence threshold and then subject to a 5% confidence threshold. We found that whilst all examples would be predicted under no confidence threshold, only 90% of the test set could be confidently predicted when the 5% confidence threshold was applied. This means that 262 examples were being assigned to a given class with only <5% difference between the top two probability estimates. Running models with very low confidence thresholds suggested that some of the correct answers that the model outputs when no thresholds are implemented turn out to be lucky guesses. This sensitivity implies that the performance of models with no minimal confidence restriction can rapidly decrease when faced with noisier datasets. Thus, we conclude that confidence threshold implementation is an efficient way to improve the robustness and reliability of a model.
We tested the effect of increasing confidence thresholds on the fraction of test examples that a model can predict with confidence and the corresponding accuracy. The model was used to predict HBND for the complete test set under different confidence thresholds. The results are shown in Fig. 4. For each confidence threshold used, there is a pair of red and blue dots representing the percentage of test examples that were predicted with confidence and corresponding prediction accuracy. Generally, as the confidence threshold increases, the accuracy of confident estimations also increases and does improve considerably. Conversely, the percentage of test examples that the model is able to predict with confidence drops rapidly. For example, whilst the absence of a confidence restriction allowed the model to predict the complete test set with an accuracy of 59%, a 30% threshold enabled the model to output predictions for 34% of the test set with an accuracy of 73%, or for 8% of the test set with an accuracy of 89% when the threshold was increased to 60%.
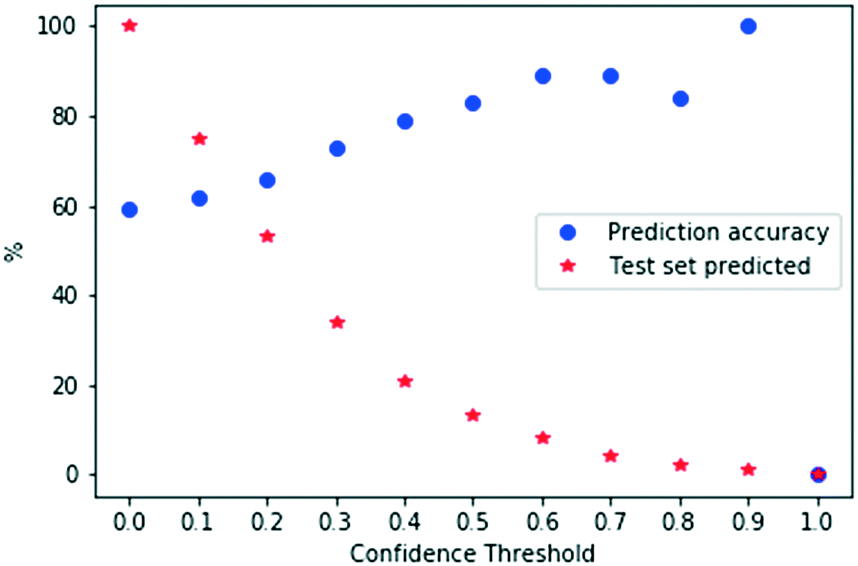 | ||
| Fig. 4 Effect of confidence thresholds on the percentage of the test set predicted with confidence and correspondent prediction accuracy. | ||
When the number of confident predictions gets too small, meaningful statistics about the general performance of the model cannot be derived. As shown in Fig. 4, the prediction accuracy for confidence values above 60% are obtained from small samples that are no longer a good representation of the original data distribution. In our case, 60% is the maximum confidence threshold to be adopted for the computation of a meaningful overall model performance. We stress that despite this, the confidence associated with the outputs obtained at high thresholds is still valid.
In summary, confidence thresholds make it possible to operate this model up to any achievable desired level of accuracy, however a compromise between accuracy and access to answers is required.
Finally, we use the confidence restriction to predict the test set over seven classification rounds of decreasing confidence thresholds. The idea was to feed into the classification round all the test examples that the model was not able to confidently predict in the previous round, so the number of confident guesses could be maximised. From the previous results (Fig. 4), it seems reasonable to start the first round with the highest confidence threshold of 60%.
We also note that the confidence threshold can be continuously decreased, as long as each round of classification still performs better than random. The results are showed in Table 1.
| Conf. threshold | Conf. predictions | Right predictions | Round accuracy |
|---|---|---|---|
| 60% (round 1) | 198 | 176 | 89% |
| 50% (round 2) | 131 | 99 | 76% |
| 40% (round 3) | 229 | 164 | 72% |
| 30% (round 4) | 323 | 206 | 64% |
| 20% (round 5) | 501 | 274 | 55% |
| 10% (round 6) | 567 | 284 | 50% |
| 0% (round 7) | 651 | 267 | 41% |
As expected, gradually relaxing the confidence restriction enables the estimation of progressively less confident new answers at each round, which increases the fraction of the test set predicted. The true value of this approach is its ability to accommodate any number of rounds and threshold values, such that the number of confident answers can be maximised whilst controlling the overall accuracy. Likewise, the setup enables the discrimination of estimation based on prediction trust. For a given round, the confidence associated to the output answers is known to lie between the confidence threshold that the current and previous round were subjected to. Moreover, the possibility of fine tuning the confidence threshold step between rounds allows one to increase the discrimination between different levels of prediction trust. Ultimately, it becomes possible to quantify the prediction trust associated with each prediction.
In conclusion, we believe that the confidence restriction tool offers the possibility of tailoring the performance of a given probability-generating classification model to the risk and cost requirements of each project.
Experimental
Software and databases
Crystal structure information was extracted from the Cambridge Structural Database (2019 release) using the CSD Python API (v.2.3.0).22 Hydrogen bond network dimensionalities were calculated as described in the Data collection section. Molecular descriptors for each molecule were calculated using the RDkit cheminformatics package (2019.03.4).23 Data manipulation was handled by the Pandas (v.0.24.2) and NumPy (v.1.17.2) packages. Machine learning classifiers, hyperparameter optimisation routines, model performance metrics, confusion matrix and learning curves were implemented using the Scikit-learn (v.0.21.2) and MatPlotlib (v.3.1.1) packages. All implementations were executed in Jupyter Lab (v.1.0.2) using Python version v.3.7.Data collection
The wealth of crystallographic information stored in the Cambridge Structural Database (CSD) was exploited to provide us with a set of molecules and corresponding crystal information, from which network dimensionalities and molecular descriptors could be accurately calculated.The CSD was searched for all organic crystal structures of a single chemical component, excluding any metals, salts, and ions, as these present additional challenges11 that won't be addressed in this study. Entries with disorder, errors or incomplete information about crystal atomic coordinates or hydrogen bonds were discarded, as they would not provide enough information for accurate network dimensionality calculation. Of these, molecules with more than one crystal structure submitted to the database were removed. This step removes conflicting data where the compound is polymorphic and its different crystal arrangements are reported to have different network dimensionalities.17 Accounting for this scenario would result in a multi label classification task that will not be covered in this paper. In a few other cases, different submissions of the same crystal were calculated to have different network dimensionalities, which may relate to the quality of crystal data and sensitivity of the dimensionality calculation tool on the definition of a hydrogen bond interaction.
All entries meeting the above search criteria were subject to hydrogen bond network dimensionality and numerical descriptor calculation. Label assignment was based on a modification to the method of Bryant et al.17 Dimensionality was calculated through the computation of the square roots of the eigenvalues of the covariance matrix of the atomic coordinates of the supramolecular structures that resulted from two different expansions of the network, through hydrogen bond intermolecular interactions, using methods from the CSD Python API. Ratios for each dimension before and after the expansion were calculated, to deduce the number of directions in which the network grew. One hundred and fifteen descriptors of two and lower dimensions were calculated for each molecule using the RDkit package. The full list of descriptors can be found in the ESI‡ A.
Data pre-processing
Data processing included cleansing, scaling and class size balancing. During cleansing, entries containing molecular descriptor with infinite or missing values were removed because they cannot be handled by the statistical methods to be used. Likewise, two descriptors that were constant across all examples were discarded, as they add no relevant information for class separation. All remaining descriptors were scaled to zero mean and unit variance, promoting similar feature contribution in classifiers that operate based on distance metrics between data points. Class size balance was achieved by an under-sampling procedure, which consists of reducing over-represented classes to the size of the smallest class by random sampling. A class balancing report that allows comparison of the distribution of a given descriptor across different datasets was implemented to check that each class sample is representative of the pool that it was drawn from.Data visualization
The balanced datasets were subject to t-Distributed Stochastic Neighbour Embedding (t-SNE), a nonlinear dimensionality reduction technique that embeds high dimensional datasets into two dimensions for visualization purposes.24The method has two main hyperparameters, which may greatly affect the final dataset visualization. Perplexity is responsible for the balance between conserving the local and global structure of data, whilst the learning rate controls step size of the optimisation procedure. Different hyper parameter values were tested, and although the arrangement of the points varies between projections, the general effect and overall conclusion are consistent. The visualization shown was produced with a learning rate of 10 and a perplexity of 40, which lies within the limits of 5 and 50 recommended by Hinton et al.24 Results were visualized under a colour scheme matching points to the class they belong to.
Machine learning models
The dataset was divided into a training (80%) and test set (20%). Several statistical methods were considered and a full list can be found in the ESI‡ B. Each statistical model was subject to a grid search hyperparameter optimisation under a 5-fold cross validation on the training set. The model yielding the highest mean cross validation accuracy was further considered. Model performance measures such as test set accuracies, confusion matrices, and learning curves were computed. The final model was configured to output likelihood estimates, which are returned as arrays with the probabilities that an example belongs to each of the existing classes. The assignment of examples to a final class was always derived from these arrays. Support vector machines (SVM) are maximal margin classifiers and do not directly generate probability estimates, which were nevertheless obtained using 5-fold cross-validation routines.25Better than random studies
Better than random studies were undertaken by y-scrambling,26 which randomly shuffles the labels of the test examples. The prediction accuracy on this new set was held as the random accuracy threshold, and any prediction accuracies above that was considered better than random.Learning curves
Learning curves show the evolution of model learning performance as the size of the training set increases. These plots can be used to diagnose how well the model is fitting the training data and generalising to unseen examples, as well as to derive upper bound accuracy limits given the type of data at hand. The learning curve was computed from the entire class-balanced dataset subject to a 5-fold cross validation, using hyperparameter optimised SVM method.Conclusions
Reliably predicting material properties from two and lower dimensional molecular information is a powerful strategy for the rapid assessment of compounds from any region of chemical space, bypassing resource intensive experimental work. Models like this are of interest to the pharmaceutical industry as they enable informed decisions on the most promising drug candidates in the early stages of drug development.We report a 4 class classifier that estimates the hydrogen bond network dimensionality that organic compounds may produce in a crystal structure, with an accuracy of 59% (where 25% is random). The limitations of predicting three dimensional properties from two dimensional chemical information have been discussed.
Model performance could not be improved further with the data at hand, but we demonstrate that the model's practical use could be improved by increasing the confidence of its output predictions. The confidence restriction proved efficient in adding robustness to the model by filtering marginal classification events due to noise in data, which we suggest as a good practice to be adopted for any classifier that is capable of outputting probabilities. The system further allows one to adjust the model's performance, maximize the number of confident predictions and discriminate them according to level of prediction trust. Nevertheless, a compromise between accuracy and access to answers is required for the achievement of useful results.
We anticipate that the HBND classification model may be useful to the pharmaceutical sector to support the early identification of molecules with high chances of exhibiting low plasticity levels or poor tabletability performance,17 so precautions can be taken from the beginning of the drug development pipeline. More broadly, we envisage that the confidence restriction measure may be a useful complementary tool for increasing the practical value of any probability-generating classification algorithm.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Puck Van Gerwen for starting and providing some insights on the classification task. This project was a funded collaboration between the University of Oxford, Pfizer and The Cambridge Crystallographic Data Centre. This work was supported by funding from the Engineering and Physical Sciences Research Council (EPSRC) and the Medical Research Council (MRC) [grant number EP/L016044/1].References
- Y.-C. Lo, S. E. Rensi, W. Torng and R. B. Altman, Machine learning in chemoinformatics and drug discovery, Drug Discovery Today, 2018, 23, 1538–1546 CrossRef CAS PubMed
.
- E. Hendriks,
et al., Industrial requirements for thermodynamics and transport properties, Ind. Eng. Chem. Res., 2010, 49, 11131–11141 CrossRef CAS
- R. J. Meier, A way towards reliable predictive methods for the prediction of physicochemical properties of chemicals using the group contribution and other methods, Appl. Sci., 2019, 9, 1700 CrossRef CAS
- D. Fourches, E. Muratov and A. Tropsha, Trust, but verify: On the importance of chemical structure curation in cheminformatics and QSAR modeling research, J. Chem. Inf. Model., 2010, 50, 1189–1204 CrossRef CAS PubMed
- T. Le, V. C. Epa, F. R. Burden and D. A. Winkler, Quantitative structure-property relationship modeling of diverse materials properties, Chem. Rev., 2012, 112, 2889–2919 CrossRef CAS PubMed
- Danishuddin and A. U. Khan, Descriptors and their selection methods in QSAR analysis: paradigm for drug design, Drug Discovery Today, 2016, 21, 1291–1302 CrossRef CAS PubMed
- R. P. Sheridan and S. K. Kearsley, Why do we need so many chemical similarity search methods?, Drug Discovery Today, 2002, 7, 903–911 CrossRef PubMed
- S. Dastmalchi, M. Hamzeh-Mivehroud and K. Asadpour-Zeynali, Comparison of different 2D and 3D-QSAR methods on activity prediction of histamine H3 receptor antagonists, Iran. J. Pharm. Res., 2012, 11, 97–108 CAS
- T. I. Oprea, On the information content of 2D and 3D descriptors for QSAR, J. Braz. Chem. Soc., 2002, 13, 811–815 CrossRef CAS
- C. H. Andrade, K. F. M. Pasqualoto, E. I. Ferreira and A. J. Hopfinger, 4D-QSAR: Perspectives in drug design, Molecules, 2010, 15, 3281–3294 CrossRef CAS PubMed
- A. M. Reilly,
et al., Report on the sixth blind test of organic crystal structure prediction methods, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 439–459 CrossRef CAS PubMed
- C. C. Sun and Y. H. Kiang, On the identification of slip planes in organic crystals based on attachment energy calculation, J. Pharm. Sci., 2008, 97, 3456–3461 CrossRef CAS PubMed
- V. Venkatraman, V. I. Pérez-Nueno, L. Mavridis and D. W. Ritchie, Comprehensive comparison of ligand-based virtual screening tools against the DUD data set reveals limitations of current 3D methods, J. Chem. Inf. Model., 2010, 50, 2079–2093 CrossRef CAS PubMed
- P. Raccuglia,
et al., Machine-learning-assisted materials discovery using failed experiments, Nature, 2016, 533, 73–76 CrossRef CAS PubMed
-
W. M. Czarnecki, S. Jastrzebski, I. Sieradzki and S. Podlewska, Active Learning of Compounds Activity – Towards Scientifically Sound Simulation of Drug Candidates Identification, 2015, ECML-PKDD 2015 2nd Workshop on Machine Learning in Life Sciences, 40-51 https://kudkudak.github.io/assets/pdf/publications/al.pdf Search PubMed
- M. Olivecrona, T. Blaschke, O. Engkvist and H. Chen, Molecular de-novo design through deep reinforcement learning, J. Cheminf., 2017, 9, 1–14 Search PubMed
- M. J. Bryant, A. G. P. Maloney and R. A. Sykes, Predicting mechanical properties of crystalline materials through topological analysis, CrystEngComm, 2018, 20, 2698–2704 RSC
- M. H. Shariare, F. J. J. Leusen, M. De Matas, P. York and J. Anwar, Prediction of the mechanical behaviour of crystalline solids, Pharm. Res., 2012, 29, 319–331 CrossRef CAS PubMed
- R. S. Payne, R. J. Roberts, R. C. Rowe, M. McPartlin and A. Bashal, The mechanical properties of two forms of primidone predicted from their crystal structures, Int. J. Pharm., 1996, 145, 165–173 CrossRef CAS
- S. Chattoraj, L. Shi and C. C. Sun, Understanding the relationship between crystal structure, plasticity and compaction behaviour of theophylline, methyl gallate, and their 1:1 co-crystal, CrystEngComm, 2010, 12, 2466–2472 RSC
- M. J. Bryant,
et al., Particle Informatics: Advancing Our Understanding of Particle Properties through Digital Design, Cryst. Growth Des., 2019, 19, 5258–5266 CrossRef CAS
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge structural database, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed
- RDKit: Open-source cheminformatics.
- L. Van Der Maaten and G. Hinton, Visualizing data using t-SNE, J. Mach. Learn. Res., 2008, 9, 2579–2605 Search PubMed
- T. F. Wu, C. J. Lin and R. C. Weng, Probability estimates for multi-class classification by pairwise coupling, J. Mach. Learn. Res., 2004, 5, 975–1005 Search PubMed
- C. Rücker, G. Rücker and M. Meringer, Y-randomization and its variants in QSPR/QSAR, J. Chem. Inf. Model., 2007, 2345–2357 CrossRef PubMed
Footnotes |
| † The model and confidence restriction measure codes are publicly available online. HBND model: https://github.com/APFrade/HBNDmodel. Confidence restriction: https://github.com/APFrade/ConfidenceMeasure. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ce00111b |
| This journal is © The Royal Society of Chemistry 2020 |