
Photochromic and photomodulated luminescence properties of two metal–viologen complexes constructed by a tetracarboxylate-anchored bipyridinium-based ligand†
Lin-Ke
Li
*a,
Hai-Yang
Li
a,
Ting
Li
a,
Li-Hong
Quan
a,
Jing
Xu
a,
Fu-An
Li
b and
Shuang-Quan
Zang
 *a
*a
aCollege of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou, 450001, P. R. China. E-mail: lilinke@zzu.edu.cn; zangsqzg@zzu.edu.cn
bCollege of Chemistry and Environment Engineering, Pingdingshan University, Pingdingshan, 467000, P. R. China
First published on 3rd September 2018
Abstract
In this study, two metal–viologen complexes, formulated as [Cd1.5(H2L)0.5(Cl)3(CH3OH)]n (1) and [Eu2(L)(NO3)4(HOCH2CH2OH)2]n (2), were constructed by a tetracarboxylate anchored bipyridinium-based ligand 1,1′-bis(3,5-dicarboxybenzyl)-4,4′-bipyridinium dichloride (H4L·Cl2). Single-crystal X-ray analyses revealed that complex 1 featured a 2D-layered structure, while complex 2 showed a 1D “ladder-like” chain structure. The difference in the central metal ions, anions, solvent molecules, deprotonation degree of the tetracarboxylate ligand, coordination modes and conformations of the ligand have a great influence on the final structure. As expected, incorporation of the viologen moiety to the frameworks led to the predefined photochromism: when samples 1 and 2 were exposed to UV-visible light, a reversible color change from light yellow to blue/dark blue occurred. Moreover, complex 2 exhibited the characteristic emission of Eu(III) ion in the visible region, and it also displayed an interesting reversible tunable photo-induced luminescence switching behavior: under the irradiation of UV-vis light, the luminescence emission intensity of complex 2 gradually decreased and reached 15% of the starting value after 8 min. These results may not only help to further understand the structure–photoresponse relationships of viologen-based MOFs, but also help to guide the design and synthesis of luminescent materials with photo-modulated switching ability.
Introduction
In recent years, articles reporting metal–organic framework (MOFs) materials have exponentially increased because of their wide applications in gas storage, sensing, separation, drug delivery, catalysis, magnetism, etc.1–8 Moreover, varieties of new application prospects have been developed, among which MOFs being investigated as chromic materials in response to external stimuli have attracted increasing interest. In well-defined structural MOFs, external stimuli such as light, heat, electricity, magnetism, mechanical force, solvent and water molecules induced chromic behaviors, which endowed these stimuli responsive MOF materials with enormous potential applications.9–16 These chromic MOF materials could be used in optoelectronic and photochemical devices, smart windows, memory devices, photomagnetism, data storage, inkless and erasable printing, nondestructive luminescence readout field and molecular switches,9–16 and so on.With the aim of producing efficient functional chromic MOF materials, photoactive organic linkers with specific structures are a preferred selection. For this purpose, viologen-functionalized ligands have been widely used in the self-assembly process because the ligand itself can undergo electron-transfer (ET) to produce viologen radicals under external light, heat or electrical stimuli, resulting in a photo-, thermo- or electrochromic behavior, respectively.17–20 Moreover, MOFs built using pre-modified viologen ligands could be used to design new photo-response materials, which may possess predetermined or improved physical/chemical properties because of the highly ordered arrangement of functional ligands and metal ions.
In recent years, our group has been using various aromatic carboxylate-anchored bipyridinium/viologen-based ligands to react with different metal ions to construct functional photoresponsive MOF materials.21 The incorporation of redox-active viologen groups can endow the as-synthesized MOF materials with charge transport properties as well as response to external stimuli. Moreover, the abundant oxygen atoms in the carboxylate groups and the pyridine nitrogen atoms are in favor of viologen-functionalized ligands' bonding with a variety of metal ions (which involve nearly the entire periodic table) to form the desired and fascinating structures.
Moreover, in most cases, lanthanide ions are excellent luminescent centers due to the high color purity and long lifetimes of their excited states.22 In addition, Ln-MOFs usually present excellent luminescent characteristics when strong absorbing chromophores that can stimulate fluorescence emission from lanthanide ions are incorporated as adjacent antennas or sensitizers.23 Aromatic carboxylate-anchored bipyridinium/viologen-based ligands have a large electron conjugated system, and may act as excellent “antennas” to induce the enhancement of fluorescence intensity upon coordination with lanthanide ions. Photo-response properties of the functional viologen-based ligand in combination with the excellent luminescence of Ln(III) complexes may result in MOF materials with multi-functional properties.
Therefore, under the guidance of these considerations, using a tetracarboxylate anchored bipyridinium-based ligand 1,1′-bis(3,5-dicarboxybenzyl)-4,4′-bipyridinium dichloride (H4L·Cl2) to react with transition and lanthanide metal ions, two metal–viologen MOFs, formulated as [Cd1.5(H2L)0.5(Cl)3(CH3OH)]n (1) and [Eu2(L)(NO3)4(HOCH2CH2OH)2]n (2), were prepared. Furthermore, the reversible photochromism of complexes 1 and 2 as well as the solid-state luminescence and photo-modulated luminescence switching properties of complex 2 were investigated. Additionally, powder X-ray diffraction (PXRD) analyses, infrared spectroscopy (IR) analyses, elemental analyses, thermogravimetric analyses (TGA) and electron-spin resonance spectroscopy (ESR) analyses of these MOFs have been performed.
Experimental
Materials and methods
Ligand 1,1′-bis(3,5-dicarboxybenzyl)-4,4′-bipyridinium dichloride (H4L·Cl2) (Scheme 1) was synthesized according to a literature procedure with some modifications.24 All other commercially available reagents were used without further purification. Elemental analyses for C, H and N were performed using a Perkin-Elmer 240 elemental analyzer. IR spectra were recorded with KBr pellets in the range of 4000–400 cm−1 on a Bruker VECTOR 22 spectrometer. Thermogravimetric analyses were performed using a TA Q50 thermal analyzer from room temperature to 800 °C with a heating rate of 10 °C min−1 under nitrogen flow. Powder X-ray diffraction (PXRD) patterns for complexes 1 and 2 were recorded on a Rigaku D/Max-3B diffractometer (Cu Kα, λ = 1.5418 Å) in the 2θ range of 5–50°. Crushed single crystalline powder samples were prepared by crushing the crystals and scanning from 5° to 50° with a step of 0.1° s−1. Solid UV-visible spectrum was obtained in the 200–800 nm range on a JASCOUVIDEC-660 spectrophotometer. Electron-spin resonance (ESR) signals were recorded on a Brucker A300 spectrometer at room temperature.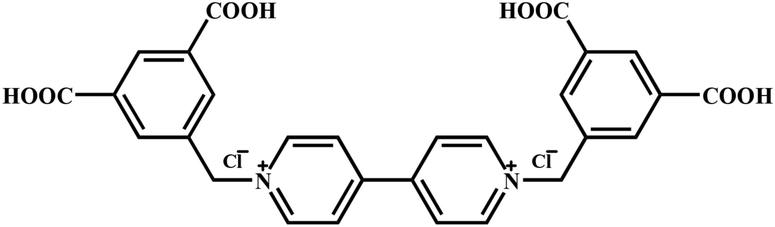 | ||
| Scheme 1 The ligand 1,1′-bis(3,5-dicarboxybenzyl)-4,4′-bipyridinium dichloride (H4L·Cl2). | ||
Preparation
Results and discussion
Crystal structure of [Cd1.5(H2L)0.5(Cl)3(CH3OH)]n (1)
Complex 1 crystallizes in a triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and features a 2D-layered structure. The asymmetric unit of 1 consists of one and a half independent Cd(II) cations (the occupancy is 1 for Cd1 and 0.5 for Cd2), one half of an H2L ligand, three coordinated Cl− ions and one coordinated CH3OH molecule.
and features a 2D-layered structure. The asymmetric unit of 1 consists of one and a half independent Cd(II) cations (the occupancy is 1 for Cd1 and 0.5 for Cd2), one half of an H2L ligand, three coordinated Cl− ions and one coordinated CH3OH molecule.
As shown in Fig. 1, in the crystal structure, both Cd(II) centers are six-coordinated, but in different coordination environments. Each Cd1 ion is coordinated by two carboxylate oxygen atoms (O1 and O4#1) from two H2L ligands in different coordination modes (one is in monodentate coordination mode and the other is in bridging mode) and four Cl− ions (Cl1, Cl2, Cl3 and Cl1#2), among which Cl1, Cl3 and Cl1#2 take part in the coordination in bridging mode, while Cl2 is in monodentate mode, forming a slightly distorted octahedral geometry (Fig. 1). Cl1, Cl2, Cl1#2 and O4#1 compose the equatorial plane (the mean derivation from the ideal CdCl3O plane is 0.0333 Å), while Cl2 and O1 exist on the axial positions; the axial angle is 171.37(8)°. The resulting CdO2Cl4 octahedron exhibits Cd1–O bond lengths of 2.300(3) and 2.442(3) Å, and Cd1–Cl bond distances in the range of 2.5071(12)–2.7271(11) Å (Table S2†). For Cd2, the CdO4Cl2 core shows a nearly perfect octahedral geometry. The coordination is provided by two oxygen atoms (O3#1 and O3#5) from two different H2L ligands in bidentate bridging mode, two oxygen atoms (O5 and O5#4) from coordinated CH3OH molecules and two bridging Cl− ions (Cl3 and Cl3#4), as shown in Fig. 1. The four oxygen atoms compose the equatorial plane (the mean derivation from the ideal CdO4 plane is 0.000 Å), and the Cd–O bond distances 2.264(3) and 2.385(3) Å. The O–Cd2–O bond angles are nearly 90° or 180°. The other two Cl− ions exist on the axial positions, and the axial angle is 180°. The Cd2–Cl bond length is 2.6385(12) Å and the Cl–Cd2–Cl bond angle is 180° (Table S2†). These values are all comparable to the other reported values of Cd(II)–viologen complexes.27
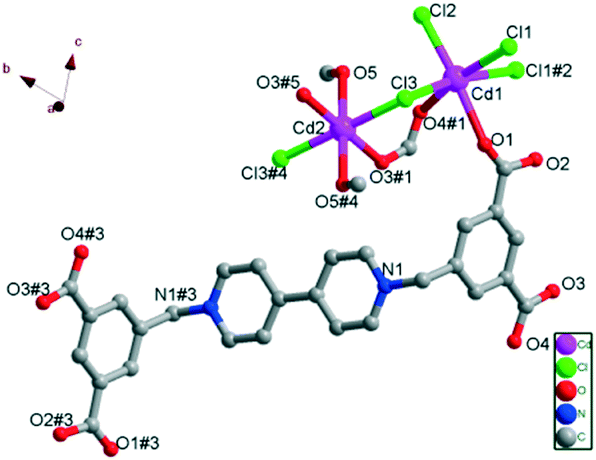 | ||
| Fig. 1 Perspective view of the coordination environment of Cd(II) ions in 1 (hydrogen atoms are omitted for clarity). Symmetry codes: #1: −x + 2, −y, −z + 1; #2: −x + 1, −y, −z + 2; #3: −x + 2, −y + 1, −z; #4: −x + 1, −y + 1, −z + 1; #5: x − 1, y + 1, z. | ||
As for the ligand H4L·Cl2, Wriedt et al. had used the ligand H4bdcbpyBr2·2H2O (1,1′-bis(3,5-dicarboxybenzyl)-4,4′-bipyridinium dibromide dihydrate solvate) to construct two isostructural MOFs {[M-(bdcbpy)(OH2)4]·4H2O}n (M = Mn and Ni), in which the ligand H4bdcbpyBr2, was fully deprotonated in the framework, yielding the anionic ligand bdcbpy2− to coordinate with the metal ions and counterbalance the M2+ metal clusters in MOFs.24 In this study, complex 1 possesses a partially deprotonated H4L·Cl2 ligand, in which only two protons (H+) of the carboxylate groups are removed, yielding the neutral ligand H2L bearing no charge. The H2L ligand presents trans-conformation. Furthermore, one H2L ligand bridges with six Cd(II) ions in two types of coordination modes: μ2-η1:η1 mode and μ1-η1:η0 mode. The O atoms connect with Cd2 only in the μ2-η1:η1 mode. Cd1 and Cd2 atoms are linked by one bridging carboxylate group from one H2L ligand and one bridging Cl− ion, forming a Cd3 unit (Cd1Cd2Cd1). The Cd3 units are repeated along the chain, and di-Cl-bridges connect the repeating Cd3 units. These Cd3 units are located nearly on a straight line. The adjacent Cd⋯Cd distances in one line are 4.0294(4) Å and 4.2054(4) Å. In addition, these two types of intervals are alternatively arranged along the line, with the adjacent Cd⋯Cd⋯Cd angles of 173.45(1)° and 180.00(0)°. All the Cd(II) ions are in a strict plane, forming the skeleton of the 2-D layer along the bc plane (Fig. 2).
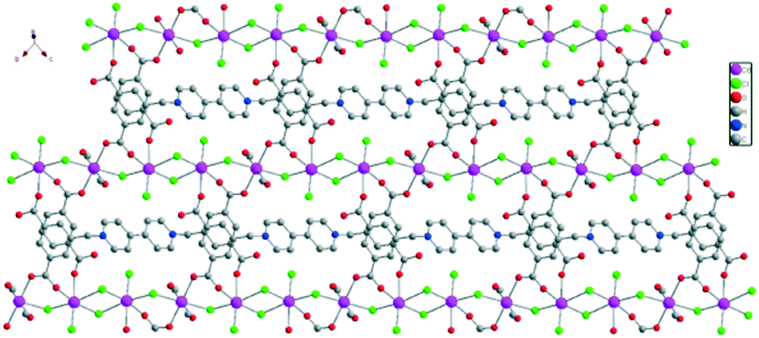 | ||
| Fig. 2 The 2D layered structure of complex 1. All hydrogen atoms are omitted for clarity. | ||
In the H2L ligand, the two pyridinium rings of the same ligand are also in a nearly ideal plane (mean deviation from the ideal plane is 0.0076 Å), and the two aromatic carboxylate moieties have the same included angles with the central pyridinium rings (111.26°) but towards different directions. The 3,5-dicarboxybenzyl units are significantly twisted from the neighboring pyridinium rings with dihedral angles of 75.63°; hence, the ligand H2L presents trans-conformation.
The adjacent layers are connected by C–H⋯Cl hydrogen bond interactions (H bond distance is 3.594 Å and the bond angle is 145.28°). Moreover, the adjacent aromatic carboxylate rings in one layer are parallel to each other along the a direction, and the nearest centroid to centroid distance is 3.785 Å.
Depending on the hydrogen bonding interactions and intermolecular forces, the 2D layered structure is extended into the 3D solid-state packed structure (Fig. 3).
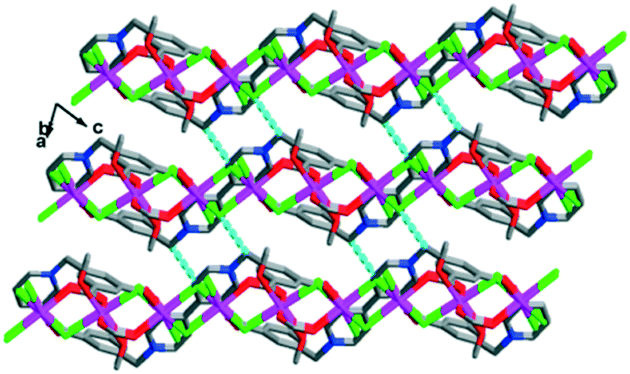 | ||
| Fig. 3 3D solid-state packed structure of complex 1 with the existence of C–H⋯Cl hydrogen bonds between the close packed layers. | ||
Crystal structure of complex [Eu(L)0.5(NO3)2(HOCH2CH2OH)]n (2)
Single-crystal X-ray diffraction analysis revealed that complex 2 crystallized in the triclinic space group P and featured a 1D “ladder-like” chain structure. As shown in Fig. 4, the asymmetric unit of 2 has one Eu(III) ion, one half of a L2− ligand, two coordinated nitrate ions and one coordinated glycol molecule. Eu1 is ten-coordinated to four oxygen atoms of two nitrate ions in η2-chelation mode, four oxygen atoms of two carboxylate groups in η2-chelation mode, and two oxygen atoms from one glycol molecule also in η2-chelation mode. The Eu–O bond lengths are in the range of 2.431(7) to 2.710(8) Å, and the bond angles around the Eu(III) center are in the range of 93.99(9)° to 129.20(9)° (Table S2†), which are comparable to those of other reported Eu(III)–oxygen complexes.
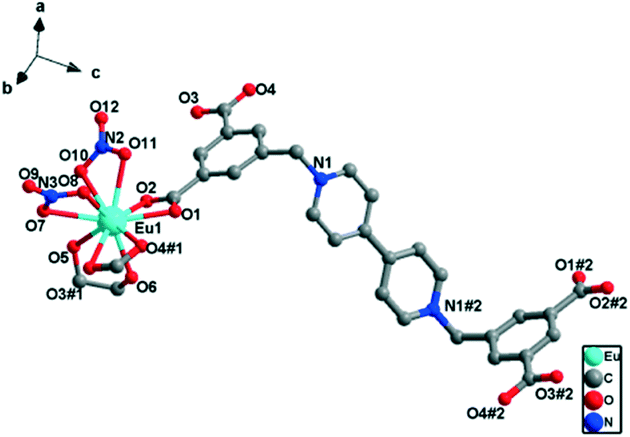 | ||
| Fig. 4 Perspective view of the coordination environment of the Eu(III) ion in 2 (hydrogen atoms are omitted for clarity). Symmetry codes: #1: x, y + 1, z, #2: −x, 1 − y, 3 − z. | ||
The coordination geometry of the completely deprotonated ligand L2− was greatly different from that of a recently reported 3D Eu(III) framework constructed by the same ligand, viz., [Eu2(OH)2(H2O)2L]Cl2·8H2O,28 in which the tetracarboxylate ligand adopts a U-shaped conformation with each carboxylate in the μ-η2:η1 coordination mode while chelating a metal ion and bridging another metal ion through a μ-O atom. However, in this case, the ligand L2− still adopts the Z-shaped trans-conformation similar to that in complex 1 and participates in coordinating in μ4-η2:η2:η2:η2 mode to connect the adjacent four Eu(III) ions to form a 1D chain along the b direction (Fig. 5). The four Eu(III) ions connected by the same L2− ligand form a parallelogram, with Eu⋯Eu lengths of 9.909 × 17.891 Å2. All the pyridinium rings and aromatic carboxylate rings in the same line are parallel to each other along the b direction. The two pyridinium rings of the same ligand are in a nearly ideal plane (mean deviation from the ideal plane is 0.0118 Å), and the two aromatic carboxylate moieties have the same included angles with the central pyridinium rings (108.7(8)°) but towards different directions, forming the Z shaped conformation (Fig. 5). The 3,5-dicarboxybenzyl units are significantly twisted from the neighboring pyridinium rings with dihedral angles of 73.46°, which is close to that of the former two complexes. The adjacent chains along the c direction are interlaced and parallel to each other, with the nearest centroid to centroid distance between the aromatic carboxylates being 3.940 Å, indicating the existence of weak face-to-face aromatic π⋯π interactions. These weak π⋯π interactions closely pack the 1D chains to form 2D layers (Fig. 5).
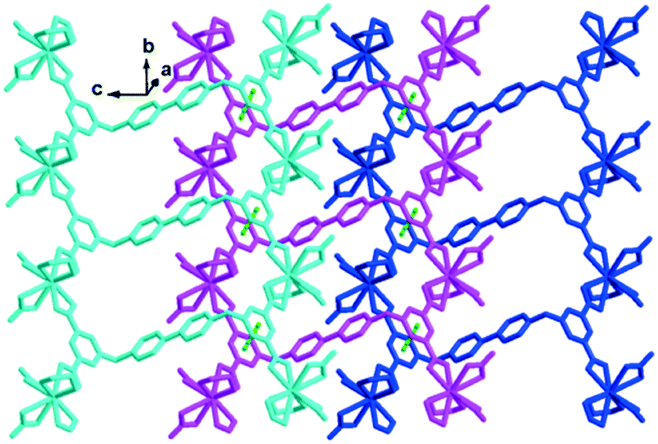 | ||
| Fig. 5 The 2D layered structure of complex 2 formed by weak π⋯π interactions (green dotted line). All hydrogen atoms are omitted for clarity. | ||
Furthermore, the weak π⋯π interactions and weak intermolecular forces tightly connect the 2D layers to form the 3D solid-state structure (Fig. 6).
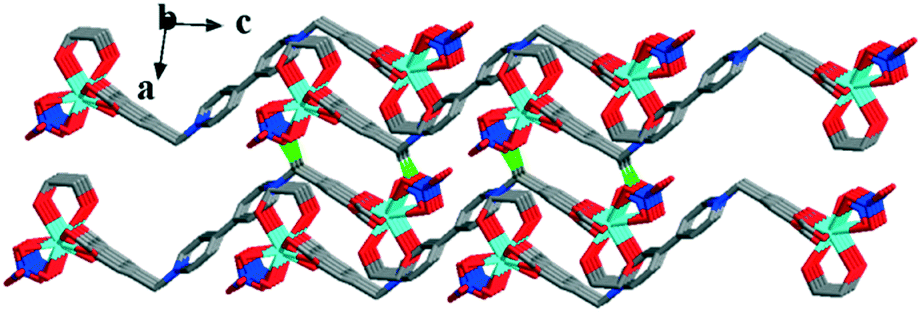 | ||
| Fig. 6 The solid-state structure of complex 2 formed by hydrogen bonds and weak π⋯π interactions. | ||
Thermostability, PXRD patterns and IR
Powder X-ray diffraction (PXRD), thermogravimetric analysis (TGA) and IR (infrared) spectroscopy patterns before and after light irradiation were obtained to confirm the stability of these frameworks (ESI†).TGA measurements of complexes 1 and 2 were performed from room temperature to 800 °C under nitrogen atmosphere to evaluate the thermal stability of the two complexes (Fig. S1†). For complex 1, upon heating, the first experimental weight loss of 5.46% from 220 to 255 °C roughly corresponds to the release of two methanol molecules (calculated 5.69%). Upon further heating, after a plateau, the weight loss corresponds to the decomposition of organic moieties in the range of 315 to 700 °C. In case of complex 2, the framework remained stable up to 225 °C and then, the two-step weight loss occurred from 225 to 396 °C, corresponding to the release of two coordinated glycol molecules and four coordinated nitrate anions. The subsequent weight loss is ascribed to the decomposition of the organic moiety.
The phase purity of the two complexes has been investigated by X-ray diffraction. As shown in Fig. S2 (ESI†), the peak positions of theoretical and experimental as well as the PXRD patterns before and after the irradiation agree well with each other, indicating the high purity and homogeneity of these complexes; also, this result showed that the structures are maintained during photochromism.
IR (Fig. S3, ESI†) spectra of complexes 1 and 2 before and after the irradiation as well as after thermal bleaching also coincided well, indicating that the framework was maintained before and after the treatment. The results also indicated that the photochromism was not caused by isomerization or photolysis. The photochromic processes of complexes 1 and 2 could be probably ascribed to the electron-transfer and the photo-induced generation of viologen radicals.
Photochromic properties
Complexes 1 and 2 were photosensitive, giving a visible color change within a few minutes upon the exposure to sunlight or the xenon lamp (300 W) in air at room temperature. Complex 1 turned from pale yellow to blue, and the coloration achieved complete saturation after irradiation for 15 minutes (Fig. 7a). Photo-induced blue crystals (1-L) regained the pale yellow color after being kept in the dark for about two days under ambient conditions or in 20 min by heating at 120 °C in air. The color of complex 2 changed from pale yellow to dark blue, and the coloration achieved complete saturation after irradiation for 10 minutes (Fig. 7b). In addition, the photo-induced product (2-L) could regain the light yellow color when maintained in the dark for one day under ambient conditions or in about 15 min by heating at 120 °C in air. Such reversible color transformations could be repeated for several continuous cycles without a noticeable color loss, indicating good reversible properties of the two complexes.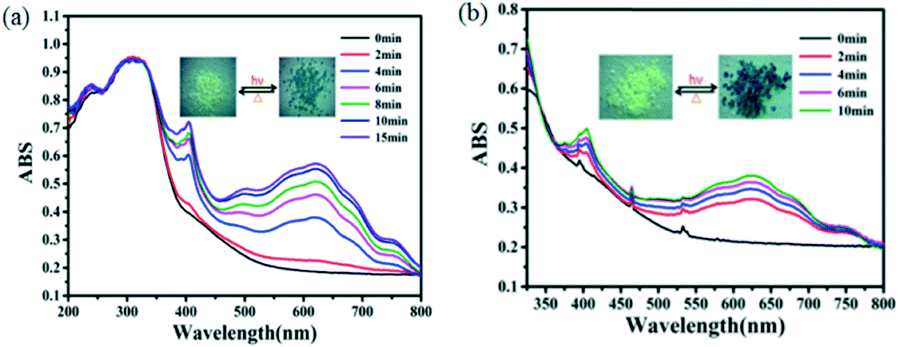 | ||
| Fig. 7 UV-vis spectra and photographs showing the photochromic behaviors of complexes 1 (a) and 2 (b), the inset is the colour change of the complexes 1 and 2 before and after irradiation. | ||
UV-vis reflectance spectra were recorded before and after illumination (Fig. 7). From the figure, we can observe that the photoproducts exhibited two characteristic bands at 403 nm and 612 nm for complex 1 and 404 nm and 625 nm for complex 2, which increase gradually with the duration of irradiation. These absorption peaks should be related to π–π* and n–π* transitions of the conjugated ligand L2−.
After irradiation, ESR spectra displayed strong and sharp signals at g = 2.0018 and 2.0019 for 1 and 2, respectively (Fig. 8), which are close to that of a free electron (2.0023) and can be assigned to radicals.20,21,29 Furthermore, the intensities of the signals of the complexes drastically increased with the irradiation time, confirming the formation of radicals. These features resemble those of photogenerated viologen radicals.20,21,29 Thus, the photochromic behaviors of complexes 1 and 2 could be ascribed to the photoinduced electron transfer and the generation of free viologen radicals.
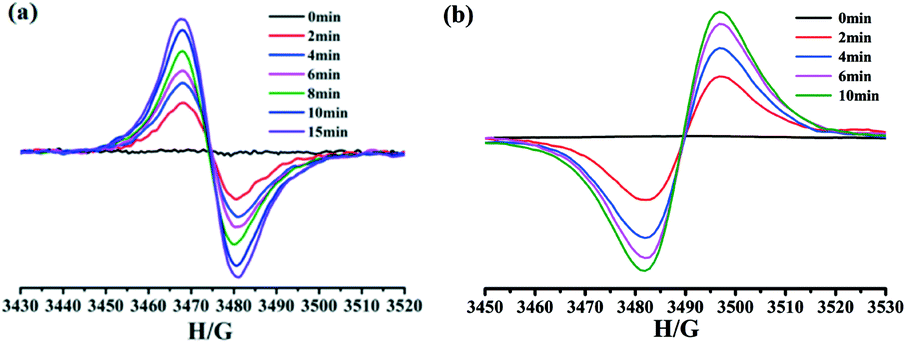 | ||
| Fig. 8 The ESR spectra for complexes 1 (a) and 2 (b) before and after the irradiation in the solid state at room temperature. | ||
The solid-state UV-vis reflectance spectrum of the ligand H4L·Cl2 was also recorded for comparison (Fig. S4†). It only shows one peak in the ultraviolet region, and the ligand itself has no photochromic property. Thus, the photochromic behaviors of the complexes are ascribed to the influence of the crystal structure.
In an attempt to gain an in-depth understanding of the influence of the structure on the photochromic behaviors, the detailed structural analyses of the complexes were performed carefully. As mentioned in previous reports, for viologen-based MOFs, several factors such as the distance between the donor and the acceptor, the orientation between the electron donor and the acceptor, the packing type, hydrogen bonding interactions, C–H⋯π interactions,17–21,24,29 as well as halide anions⋯acceptor interaction30 may have a great influence on the final photochromic behaviors. In case of the two complexes, these are still the main factors to be considered. For complex 1, the shortest distance between the carboxylate oxygen atom and the N+ of the pyridinium ring is 3.387 Å, the shortest distance between the Cl atom and the N+ of the pyridinium ring is 3.421 Å and the δ angles between the donor-N+ line and the normal of the pyridinium plane are about 22.34° and 67.64°, respectively (Fig. S5†). Furthermore, the existence of weak π–π stacking interactions between the aromatic rings in neighboring bipyridinium ligands and the hydrogen bonding interactions also are favorable for electron transfer between the donor and the acceptor. For complex 2, as the central metal ion Eu(III) is a lanthanide metal ion, it instinctively exhibits high oxophilicity (hard acid hard base interaction)31,32 and usually coordinates strongly to the oxygen donor. Thus, the abundant O atoms in the coordination environment, originating from carboxylate groups and the coordinated nitrate anions, provided much more donor⋯acceptor electron transfer pathways. Furthermore, the shortest O⋯N distance is 3.456 Å, while the other O⋯N distances are 3.664, 3.708 and 3.779 Å, respectively (Fig. S5†). The δ angles between the donor-N+ line and the normal of the pyridinium plane are about 67.64°, 22.34°, 24.72° and 25.24°. These distances and angles are all favorable for electron transfer. At the same time, weak offset π⋯π interactions between the aromatic carboxylates also contributed to the photochromic behavior of complex 2.
Photoluminescence properties
Lanthanide MOFs have usually shown good luminescence properties in terms of their high color purity. Hence, we also investigated the ligand-assisted photoluminescence properties of solid-state samples of the Eu(III) complex at room temperature. Moreover, for metal–viologen complexes, photoluminescence modulation property has also been observed and it has attracted considerable attention in recently reported chromic MOF materials.17,20,33The solid-state luminescent property of ligand H4L·Cl2 was characterized by recording a UV-vis reflectance spectrum (Fig. S4†). It is observed that in the spectral region from 230 to 400 nm, ligand H4L·Cl2 exhibits one intense broad excitation band centered at about 300 nm, with a rapidly decreasing tail in the visible light region.
The solid-state photoluminescence behavior of complex 2 before and after irradiation was also investigated. Excitation of the as-synthesized solid of complex 2 at room temperature resulted in peaks at 361, 375, 395, 414, 464 and 532 nm, which were ascribed to the electron transitions of 7F0 → 5D4, 7F0 → 5L7, 7F0 → 5L6, 7F0 → 5D3, 7F0 → 5D2 and 7F1 → 5D1, respectively (Fig. 9a).
 | ||
| Fig. 9 (a) Solid-state excitation spectrum of complex 2 at room temperature. (b) The luminescence emission spectral changes (λex = 395 nm) of complex 2 with the time of irradiation. | ||
Under the excitation of 395 nm at room temperature, the typical red emission of the Eu(III) ion in the visible region was detected for complex 2 (Fig. 9b), and the five sharp peaks appearing at 578, 586, 616, 646 and 690 nm correspond to the characteristic f–f transitions of 5D0 → 7FJ (J = 0–4) of Eu(III) ions. Similar to the other Eu(III) complexes, the electric dipole 5D0 → 7F2 transition is the strongest among all the transitions, and it is responsible for the bright red luminescence of the complex. Moreover, since the fluorescence intensity ratio of 5D0 → 7F2 to 5D0 → 7F1 is sensitive to the structural change in the vicinity of Eu(III) ions, the intensity ratio (ca. 7) of I(5D0 → 7F2)/I(5D0 → 7F1) indicated the low symmetry of the Eu(III) site in complex 2.34 This is consistent with the crystal structure analysis.
It has been reported that viologen radicals are typical luminescence quenchers.35 Under the irradiation of UV light, the luminescence emission intensity (395 nm) of complex 2 exhibited a gradual decrease with time and reaches 15% of the starting value after 8 min (as shown in Fig. 9b). The photoswitching process can be repeated several times without a noticeable loss in luminescence intensity (Fig. 10a). As for the reason of luminescent switching, we can surmise a conclusion from Fig. 10b: there exists a spectral overlap between the emission band of the initial sample and the broad UV-vis absorption band of the colored sample, which can result in the energy transfer from the excited luminescent center to the L2− free radical. Thus, the luminescence switching behaviors of complex 2 were induced. This photomodulated luminescence switching behavior combined with photochromism is an interesting property, which may result in more and broad application prospects for this type of metal–viologen complexes in the photoactivity field.
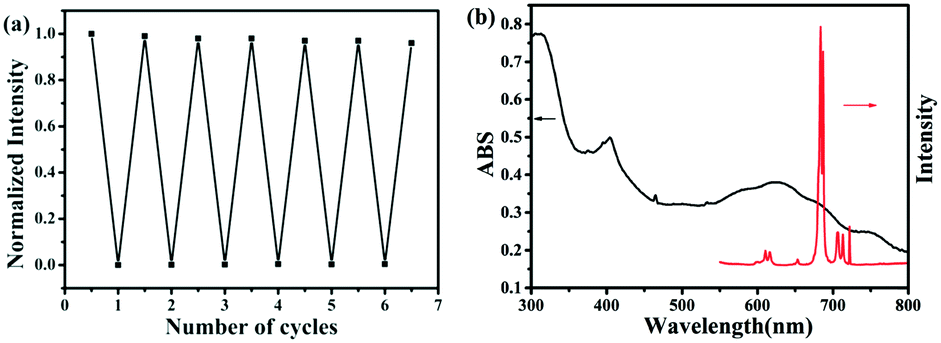 | ||
| Fig. 10 (a) The repeated switching of luminescence emission of complex 2 between the initial and colored states based on photochromic modulation. (b) The luminescence emission spectra of complex 2 and the ligand in the solid-state at room temperature. | ||
Conclusions
In summary, utilizing an aromatic polycarboxylate-anchored bipyridinium-based ligand to react with different metal ions, two metal–viologen complexes with different structures have been successfully synthesized. The central metal ions, deprotonation degree of polycarboxylate groups, and the coordination geometry of viologen-based ligands have a great impact on the final structure of MOFs, and further result in their distinct photochromic behaviors. Moreover, when coordinated with the luminescent lanthanide Eu(III) metal ion, the final complex exhibits characteristic emission of Eu(III) ion in the visible region, and the viologen-based ligand could act as an effective “antenna” to induce the enhancement of fluorescence intensity. The existence of the viologen moiety induced the photomodulated luminescence change in the Eu(III)–viologen MOF. The structural analyses of these complexes revealed that the electron transfer between the carboxylate-O donor and the pyridinium-N acceptor is the dominating factor for photochromic behaviors. Moreover, the abundant O atoms in the tetracarboxylate anchored bipyridinium-based ligand provide much more electron transfer pathways, which increase the possibility for the effective electron transfer to happen, thus resulting in the occurrence of the photochromic behaviour.Systematic investigations on this type of MOF materials are still underway to search for more chromic materials. The results obtained in this paper can not only help to understand the structure–photosensitivity relationships of viologen-based MOF systems, but also guide the design and synthesis of more luminescent materials with photomodulated luminescence switching properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 21371153 and 21801227), Key Scientific Research Project Plan in Colleges and Universities of Henan Province (16A150045) and Scientific and technological project in Henan Province (162102410070) and Zhengzhou University student innovation and entrepreneurship training program (2018cxcy006).Notes and references
- L. Ungur, S. Y. Lin, J. Tang and L. F. Chibotaru, Chem. Soc. Rev., 2014, 43, 6894–6905 RSC
.
- A. Bianchi, E. Delgado-Pinar, E. García-España, C. Giorgi and F. Pina, Coord. Chem. Rev., 2014, 260, 156–215 CrossRef
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308–2322 RSC
- H. Wang, J. Xu, D.-S. Zhang, Q. Chen, R.-M. Wen, Z. Chang and X.-H. Bu, Angew. Chem., Int. Ed., 2015, 54, 5966–5970 CrossRef PubMed
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef PubMed
- D. Cunha, M. Ben Yahia, S. Hall, S. R. Miller, H. Chevreau, E. Elkaïm, G. Maurin, P. Horcajada and C. Serre, Chem. Mater., 2013, 25, 2767–2776 CrossRef
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkovc and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC
- P. Dechambenoit and J. R. Long, Chem. Soc. Rev., 2011, 40, 3249–3265 RSC
- J. T. Sampanthar, K. G. Neoh, S. W. Ng, E. T. Kang and K. L. Tan, Adv. Mater., 2000, 12, 1536–1539 CrossRef
- V. Stavila, A. A. Talin and M. D. Allendorf, Chem. Soc. Rev., 2014, 43, 5994–6010 RSC
- S. Kawata and Y. Kawata, Chem. Rev., 2000, 100, 1777–1788 CrossRef PubMed
- M. D. Ward, J. R. White and A. J. Bard, J. Am. Chem. Soc., 1983, 105, 27–31 CrossRef
- L.-Z. Cai, Q.-S. Chen, C.-J. Zhang, P.-X. Li, M.-S. Wang and G.-C. Guo, J. Am. Chem. Soc., 2015, 137, 10882–10885 CrossRef PubMed
- B. Garai, A. Mallick and R. Banerjee, Chem. Sci., 2016, 7, 2195–2200 RSC
- X.-D. Yang, L. Sun, C. Chen, Y.-J. Zhang and J. Zhang, Dalton Trans., 2017, 46, 4366–4372 RSC
- P.-X. Li, M.-S. Wang, M.-J. Zhang, C.-S. Lin, L.-Z. Cai, S.-P. Guo and G.-C. Guo, Angew. Chem., Int. Ed., 2014, 53, 11529–11531 CrossRef PubMed
- G. Xu, G.-C. Guo, M.-S. Wang, Z.-J. Zhang, W.-T. Chen and J.-S. Huang, Angew. Chem., Int. Ed., 2007, 46, 3249–3251 CrossRef PubMed
- C. H. Zhang, L. B. Sun, Y. Yan, H. Z. Shi, B. L. Wang, Z. Q. Liang and J. Y. Li, J. Mater. Chem. C, 2017, 5, 8999–9004 RSC
- J.-K. Sun, P. Wang, C. Chen, X.-J. Zhou, L.-M. Wu, Y.-F. Zhang and J. Zhang, Dalton Trans., 2012, 41, 13441–13446 RSC
- H.-X. Zhang, Q.-X. Yao, X.-H. Jin, Z.-F. Ju and J. Zhang, CrystEngComm, 2009, 11, 1807–1810 RSC
- H.-Y. Li, Y.-L. Wei, X.-Y. Dong, S.-Q. Zang and T. C. W. Mak, Chem. Mater., 2015, 27, 1327–1331 CrossRef
- J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC
- Y. J. Cui, Y. F. Yue, G. D. Qian and B. L. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef PubMed
- D. Aulakh, J. R. Varghese and M. Wriedt, Inorg. Chem., 2015, 54, 1756–1764 CrossRef PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 457 CrossRef
-
G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed
- J.-J. Liu, Y.-F. Guan, M.-J. Lin, C.-C. Huang and W.-X. Dai, Cryst. Growth Des., 2016, 16, 2836–2842 CrossRef
- T. Gong, P. Li, Q. Sui, J. Q. Chen, J. H. Xu and E.-Q. Gao, J. Mater. Chem. A, 2018, 6, 9236–9244 RSC
- E. M. Kosower and J. L. Cotter, J. Am. Chem. Soc., 1964, 86, 5524–5527 CrossRef
- X.-Y. Lv, M.-S. Wang, C. Yang, G.-E. Wang, S.-H. Wang, R.-G. Lin and G.-C. Guo, Inorg. Chem., 2012, 51, 4015–4019 CrossRef PubMed
- M. Watanabe, T. Nankawa, T. Yamada, T. Kimura, K. Namiki, M. Murata, H. Nishihara and S. Tachimori, Inorg. Chem., 2003, 42, 6977–6979 CrossRef PubMed
- V. Patroniak, P. N. W. Baxter, J.-M. Lehn, Z. Hnatejko and M. Kubicki, Eur. J. Inorg. Chem., 2004, 2379–2384 CrossRef
- X.-H. Jin, J.-K. Sun, X.-M. Xu, Z.-H. Li and J. Zhang, Chem. Commun., 2010, 46, 4695–4697 RSC
- S. Sivakumar, M. L. P. Reddy, A. H. Cowley and R. R. Butorac, Inorg. Chem., 2011, 50, 4882–4891 CrossRef PubMed
- A. Beneduci, S. Cospito, M. L. Deda and G. Chidichimo, Adv. Funct. Mater., 2015, 25, 1240–1247 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. CCDC Crystallographic data in CIF format, tables of selected bond distances and angles, TGA, experimental and calculated powder XRD patterns and IR spectra. CCDC reference numbers 1859387 and 1859388 for complexes 1 and 2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ce01335g |
| This journal is © The Royal Society of Chemistry 2018 |