
Halide ion recognition via chalcogen bonding in the solid state and in solution. Directionality and linearity†
Vijith
Kumar
 ,
César
Leroy
and
David L.
Bryce
*
,
César
Leroy
and
David L.
Bryce
*
Department of Chemistry and Biomolecular Sciences & Centre for Catalysis Research and Innovation, University of Ottawa, 10 Marie Curie Private, Ottawa, Ontario K1N 6N5, Canada. E-mail: dbryce@uottawa.ca; Fax: +1 613 562 5170; Tel: +1 613 562 5800 ext.2018
First published on 12th September 2018
Abstract
Group 16 chalcogen atoms may exhibit Lewis acidic σ-holes which are able to form attractive supramolecular interactions with Lewis bases via chalcogen bonds (ChB). Interest in ChB is increasing rapidly; however, the potential for anion binding has not been fully explored. Herein we report on the application of chemically robust and stable benzylic selenocynates (1 and 2) for halide ion recognition in the solid state and in solution. Single crystal X-ray structural analysis of various cocrystals reveals structurally important Se⋯X− (X = Cl, Br, I) chalcogen bonds. Changes in the 13C and 77Se chemical shifts via NMR spectroscopy show how halide ion recognition influences the local electronic environment of the selenium atoms in solution. Deviations of the interaction from the extension of the C–Se covalent bond are quantified and assessed with the aid of computational chemistry.
Introduction
In the sundry areas of supramolecular chemistry, crystal engineering, materials science, and biochemistry, noncovalent interactions are of key import for structural and functional reasons. While conventional hydrogen bonding (HB) has long been acknowledged as a vital interaction,1 halogen bonding (XB) is also now recognized as a critical component of a variety of supramolecular architectures.2 As with XB, chalcogen bonding (ChB) is a subgroup of the σ-hole interactions. ChB results from the interaction between an electrophilic region of a group 16 element (e.g., S, Se, Te) and an electron donor, and has recently emerged as a promising parallel noncovalent interaction.3 As a result of their strength and directionality, comparable to HB and XB, chalcogen bonds have recently taken on increased prominence in a range of fields including crystal engineering, organic reactivity, supramolecular self-assembly, and pharmaceuticals.4–7Benzo-selenazoles/tellurazoles,8 bithiophenes,9 tellurazole N-oxides,10 selenophtalic anhydrides,11 bistellurophene,12 and bis(benzimidazolium-selenomethyl) derivatives13 were considered as conceptually innovative motifs for the design of ChB systems. Seleno/tellurocyanides are particularly appealing candidates. They were originally identified several years ago and re-explored recently14 due to the electron-withdrawing nature of the nitrile group which promotes the formation of a strong σ-hole on the chalcogen atom along the prolongation of the NC–Se/Te bond.13 A detailed CSD survey of the previously reported crystal structures of organic selenocyanates confirmed this hypothesis, i.e., that most of the simple aliphatic and aromatic organic selenocyanates do crystallise with ChB interactions. In the absence of other Lewis bases, the Se atom interacts with the N atom of a neighbouring selenocyanate moiety.15 Recently, Fourmigué and co-workers reported that benzylic selenocyanates are strong and directional ChB donors for crystal engineering purposes, where the chain-like motifs can be tuned into 1D extended structures upon co-crystallization with 4,4-bipyridine as a ditopic ChB acceptor.16
Although substantial evidence is growing which demonstrates the existence of ChB in the solid state, solution-phase studies involving ChB are fewer. Recently, two elegant studies demonstrating ChB-assisted anion recognition with benzotelluradiazoles17a and 5-(methylchalcogeno)-1,2,3-triazole in macrocycles17b have been described. Although the heavier chalcogens (Se, Te) are expected to have enhanced ChB donor properties, their compounds can be susceptible to oxidative decomposition, thus restricting the incorporation of such elements into more complex systems and rendering difficult studies of anion binding properties. Herein, we show that benzylic selenocyanate derivatives are chemically robust and stable ChB motifs for anion recognition (Fig. 1) that can adapt their conformations to enable halide ion recognition in solution and in the solid state. We also quantify the directional relationships between the C–Se bond extension, the maximum of the molecular electrostatic potential surface, and the position of the ChB acceptor.
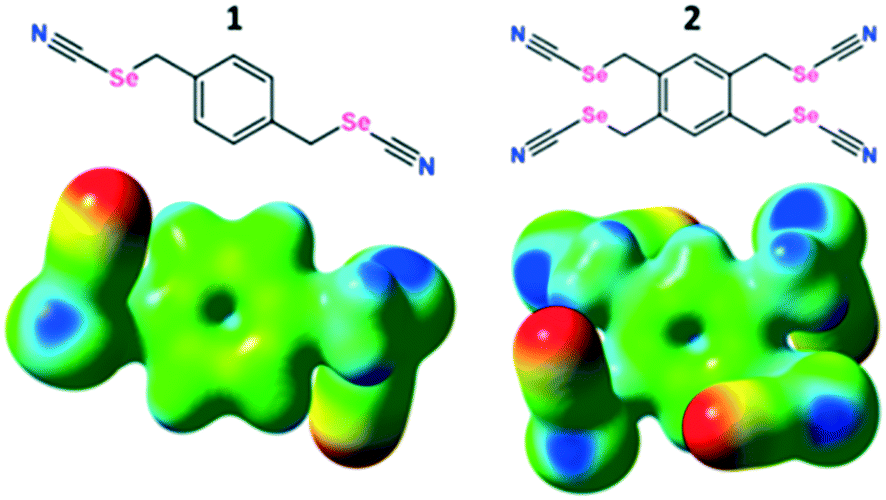 | ||
| Fig. 1 Molecular structures of the benzylic selenocyanates (top) and their MESP (bottom) with an isodensity of 0.02 a.u. (B3LYP with 6-311G**). Red indicates negative charge density, and blue positive charge density. A common scale was used to compare the surfaces visually. | ||
Results and discussion
The bis and tetrakis benzylic selenocyanates (1 and 2) are prepared from their respective benzyl bromides and potassium selenocyanates in DMF under an argon atmosphere (see Experimental).18 Structural analysis of the pure donors via single-crystal X-ray diffraction reveals that the crystal packing is mainly driven by intramolecular Se⋯N ChB interactions, resulting in a chain-like structure for 1 (ref. code POXYEH,15 ESI† Fig. S.3) and layered motifs in 2 (ref. code PEYRET,16b ESI† Fig. S.4). In the field of halogen bonding, ditopic or tritopic structures have been recently considered for anion recognition purposes due to their abilities to adapt their conformational geometries.19With this inspiration, we investigated the cocrystallisation of ChB donors 1 and 2 with tetrabutylammonium halides (iodide, bromide, and chloride). Cocrystallisation was performed via diffusion of Et2O over an acetone solution of both tectons (this produced crystals of better quality for SCXRD analysis than did simple slow evaporation from acetone); the amount of onium halide added in each case was based on the number of Se donor sites available for ChB formation (see the Experimental section for details). Complex formation is preliminarily confirmed by PXRD analysis (see Fig. 2 and ESI† S.2) and changes in the melting point of the complex in comparison with pure individual tectons. Single-crystal X-ray structural analysis of the complexes shows that ChB complexes 1·(Bu4N)Br and 1·(Bu4N)I crystallise in the monoclinic space group P21/c with Z = 4 (Table 2). Both of the structures exhibit similarity in the crystal packing, where the short and directional Se⋯I− and Se⋯Br− ChBs are largely responsible for the self-assembly of the two complementary modules. The topology of the primary network is a good example of the paradigm of the expansion of a ditopic starting module by a halide linker moiety: 1 acts as a ditopic ChB-donor while onium halides behave as strong ChB acceptors (Fig. 3, ESI† Fig. S5 and S6). The geometrical parameters describing the ChB contacts are reported in Table 1.
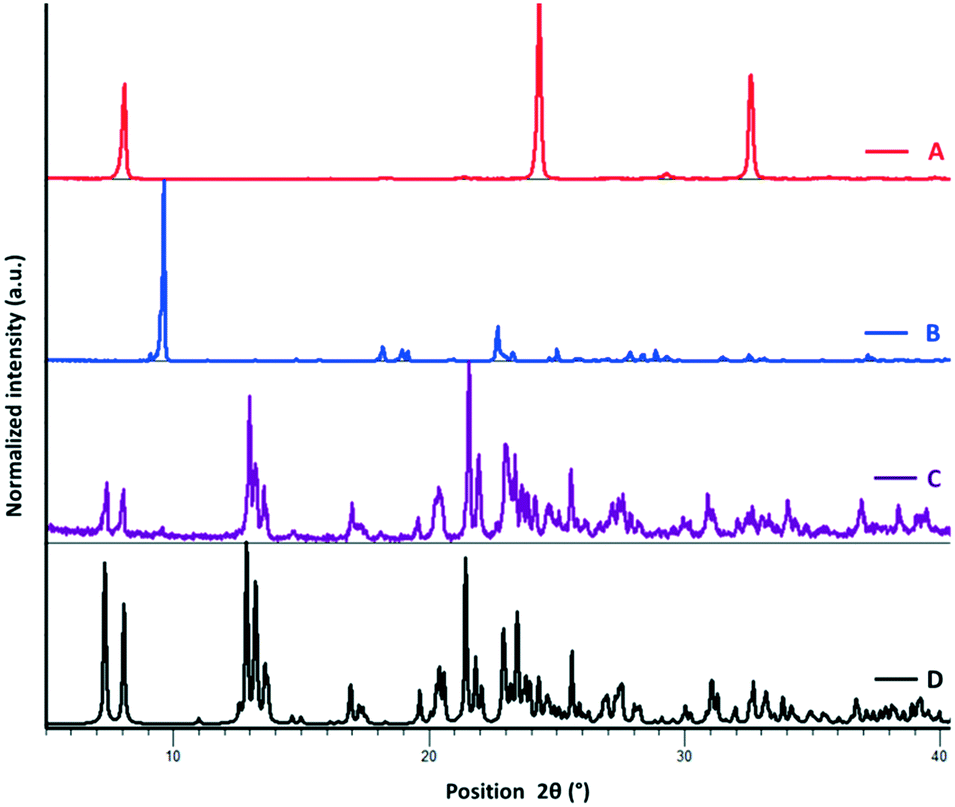 | ||
| Fig. 2 PXRD patterns of ChB donor, onium iodide, and cocrystal: experimental pattern of pure ChBD 1 (A, red); experimental pattern of pure (Bu4N)I (B, blue); experimental pattern of the 1·(Bu4N)I cocrystal obtained by solution crystallization (C, purple), simulated pattern of the 1·(Bu4N)I cocrystal obtained from the single-crystal structure (D, black). | ||
 | ||
| Fig. 3 Top: Displacement ellipsoid plots (ellipsoids are drawn at the 50% probability level) of chalcogen bonded complexes running along the crystallographic b axis, 1·(Bu4N)I (top left) and 1·(Bu4N)Br (top right); colour codes: grey, carbon; white, hydrogen; yellow, selenium; blue, nitrogen; purple, iodine; brown, bromine (dashed black lines denote ChB). Bottom: Hirshfeld surfaces plotted against dnorm for the cocrystals 1·(Bu4N)I (bottom left) and 1·(Bu4N)Br (bottom right): the red colour highlights the area of ChB formation. | ||
| Cocrystal | Se⋯X− (Å) | C–Se⋯X− (°) | Nca |
|---|---|---|---|
| a The normalized contact Nc is defined as the ratio DXY/(rX + rY), where DXY is the experimental distance between the selenium atom and halide anions and rX is the van der Waals radius for selenium (1.90 Å) and rY is the Pauling ionic radius of the halide anion Y (I−, 2.16 Å; Br−, 1.95 Å; Cl−, 1.81 Å), respectively. | |||
| 1·(Bu4N)I | 3.506(1) | 174.98(9) | 0.86 |
| 1·(Bu4N)Br | 3.249(1) | 174.40(2) | 0.84 |
| 1·(Bu4N)Cl | 3.192(2) | 174.42(3) | 0.86 |
| 2·(Bu4N)I | 3.451(1) | 171.92(1) | 0.85 |
| 3.511(1) | 170.86(1) | 0.86 | |
To test the generality and the robustness of ChB, we have challenged the ditopic ChB donor 1 with the lighter onium chloride; the cocrystal forms in the monoclinic space group I2/m with Z = 4, with both 1 and onium chloride located on inversion centers (Table 2). Several attempts were made to obtain good quality crystals for SCXRD analysis. A series of solvents and various crystallization techniques were employed (see Table S.1 in the ESI†). Slow solvent evaporation, rapid evaporation, slow cooling, vapour diffusion, and convection crystallization in a narrow tube were attempted. Even in the best cases, a soft multicrystalline material was obtained. The cocrystal has a low melting point. In addition to these problems, one of the n-butyl groups is disordered about a crystallographic mirror plane. All of these factors contribute to the woeful Rint value (ESI†). Interestingly, donor 1 adapts its conformation and the chloride anions behave as bidentate ChB acceptors (Fig. 4). The efficiency of this coordination is further evidenced by the cocrystallisation of tetrakis substituted donor 2 with onium halides. Complex 2·(Bu4N)I once again crystallises in the monoclinic space group P21/c with Z = 2, with both 2 and onium iodide located on the inversion centers, and where the iodide ion acts as a bidentate ChB acceptor and engages in short and directional Se⋯I− ChBs with the 4 selenocyanate groups (Fig. 4; ESI† Fig. S7). Unfortunately, we were unable to obtain crystals of 2·(Bu4N)Br and 2·(Bu4N)Cl suitable for SCXRD analysis; however, ChB-driven complex formation was confirmed by solution NMR spectroscopy (vide infra; ESI† S.4).
| Compound | 1·(Bu4N)I | 1·(Bu4N)Br | 1·(Bu4N)Cl | 2·(Bu4N)I |
|---|---|---|---|---|
| CCDC number | 1849844 | 1849846 | 1851129 | 1849845 |
| Empirical formula | C21H40N2SeI | C21H40BrN2Se | C26H44ClN3Se2 | C46H82I2N6Se4 |
| Molecular weight (g mol−1) | 526.41 | 479.42 | 592.01 | 1288.81 |
| Crystal size (mm3) | 0.26, 0.2, 0.14 | 0.24, 0.21, 0.15 | 0.25, 0.19, 0.12 | 0.24, 0.18, 0.17 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c | I2/m | P21/c |
| Temperature (K) | 200 | 200 | 200 | 200 |
| a (Å) | 8.059(4) | 7.8550(12) | 12.5130(13) | 12.145(4) |
| b (Å) | 14.506(6) | 14.479(2) | 14.1548(18) | 8.651(2) |
| c (Å) | 21.979(10) | 21.667(3) | 17.6517(18) | 26.550(8) |
| α (°) | 90 | 90 | 90 | 90 |
| β (°) | 90.494(11) | 90.775(3) | 101.041(7) | 96.341(7) |
| γ (°) | 90 | 90 | 90 | 90 |
| V (Å3) | 2569(20) | 2464.1(6) | 3068.6(6) | 2772.5(15) |
| Z | 4 | 4 | 4 | 2 |
| μ (mm−1) | 2.669 | 3.152 | 2.514 | 3.794 |
| R_all, R_obs | 0.045, 0.031 | 0.110, 0.057 | 0.152, 0.092 | 0.055, 0.032 |
| wR2_all, wR2_obs | 0.080, 0.075 | 0.163, 0.146 | 0.237, 0.198 | 0.073, 0.067 |
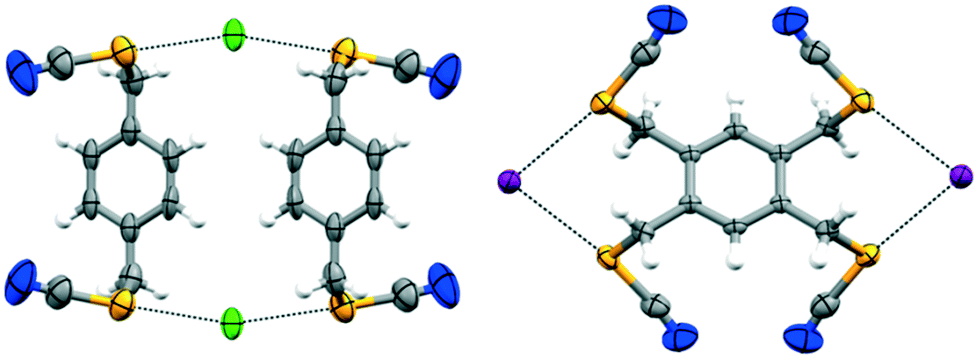 | ||
| Fig. 4 Molecular structures represented as displacement ellipsoid plots (ellipsoids are drawn at the 50% probability level) of chalcogen bonded complexes 1·(Bu4N)Cl (left) and 2·(Bu4N)I (right) show the adaptation of the geometry of the benzylic selenocyanate donors to interact with halide ions; colour codes: grey, carbon; white, hydrogen; yellow, selenium; blue, nitrogen; purple, iodine; green, chlorine (dashed black line denotes ChB). | ||
All the observed ChBs are quite short and linear (Table 1), with values similar to those reported for other chalcogen bonded systems involving selenium derivatives.16b These features confirm, once again, that the supramolecular synthon Se⋯X− is strong and reliable and that the onium halide ChB acceptor module is a very efficient building unit to promote the formation of ChB adducts. In addition to ChB, crystal packing is further stabilised by HB between the nitrile N atom and hydrogens of the onium salts' aliphatic chains in all cocrystals.
The halide anions vary in size from 119 to 206 pm (fluoride to iodide anions when octahedrally coordinated), and they can template correspondingly different self-assembled structures.20 It has been shown that the different halides can drive the formation of different structures when involved in electrostatic interactions, metal coordination, HB,21 and XB,22 and the same is noted here for ChB.
The “molecule in crystal” approach implicit in Hirshfeld surface analysis was utilized to make quantitative comparisons between contributions to crystal packing from various types of intermolecular contacts.23 This method enables the identification of individual interactions throughout the electron density weighted molecular surfaces or Hirshfeld surfaces. Hirshfeld surfaces of the cocrystals are mapped with dnorm and the electrostatic potential plotted on the Hirshfeld surface, where the ChB interactions are visualized in red (Fig. 3 bottom; ESI† S.5), depicts ChB formation in the cocrystals. The analysis carried out for pure ChB donors and cocrystals quantifies the intermolecular interactions and highlights the contributions from Se⋯X−, C–N⋯H, and other short contacts (ESI† Fig. S11).
As a preliminary gauge of the ChB donor ability, molecular electrostatic potential surfaces (MESP) were calculated for the pure ChB donors and their cocrystals. The MESP maps were visualized and traced over electron density surfaces with an isodensity of 0.02 a.u. (electron per bohr3). MESP surfaces of the pure ChB donors clearly show the presence of two electropositive regions (dark blue) on each Se atom (528.8 and 622.2 kJ mol−1 for 1 and 547.67 and 638.78 kJ mol−1 for 2 respectively); the region along the prolongation of the electron withdrawing Se–CN bond is more positive than the region along the extension of the Se–CH2 single bonds (Fig. 1 bottom, ESI† Fig. S.12, Table S.3). These positive regions, characteristic of σ-holes on the chalcogen atoms, interact with the negative MESP surface on the halide ions (red regions) functioning as electron donors (Fig. 5; ESI† Fig. S.13), individually corresponding to the Se⋯Cl−, Se⋯Br−, Se⋯I− (Table 1) chalcogen bonds differing slightly in length that increase with the size of the halide ion. In addition, the C–Se⋯X− ChB angles range from 170.86 to 174.98° (Table 1), deviating slightly from linearity, akin to XB, the sister σ-hole interaction.24 The presence of the halide induces a decrease in the VS,max on the Se atoms compared to the free donor (ESI† Table S.3).
 | ||
| Fig. 5 Electrostatic potential surface with an isodensity of 0.02 a.u. of cocrystals: 1 with (Bu4N)Br (left); 1 with (Bu4N)Cl (right) (B3LYP with 6-311G**). Red indicates negative charge density, and blue positive charge density. A common scale was used so that the surfaces can be compared visually. | ||
Having established the involvement of ChB interactions in the anion binding behaviour of selenium in the crystalline state, 13C and 77Se NMR were used to qualitatively monitor halide binding in solution (ESI† S.4). The 13C chemical shift of the methylene carbon covalently bonded to the selenium in the ChB donor can be a good indicator for the occurrence of ChB in solution. ChB causes a decrease in the chemical shift (Δδ(13C) ranging from 0.85 to 2.84 ppm) upon the formation of the C–Se⋯X− motif (Table 3). 77Se NMR is also highly sensitive to changes in the electronic and structural environment. As shown in Table 3, halide ion binding to ChB donors 1 and 2 in DMSO-d6 resulted in large changes in the 77Se NMR chemical shifts, ranging from −2.23 ppm (for 2·(Bu4N)I) to −9.56 ppm (for 2·(Bu4N)Cl). A detailed quantum chemical investigation of the origins of these shifts is beyond the scope of this communication; however, it is clear that anion binding significantly affects the electronic environment of the selenium donor atom.
| ChB donor | ChB acceptor | Δδ(77Se) | Δδ(13C) CH2 |
|---|---|---|---|
| 0.25 M solutions of ChB donors in DMSO-d6 were used. δ(77Se): 321.16 ppm, δ(13C) CH2: 32.79 ppm for 1; δ(77Se): 311.12 ppm, δ(13C) CH2: 29.62 ppm for 2. The amount of ChB acceptor added was chosen in order to form a solution containing equimolar amounts of ChB donor and acceptor atoms. | |||
| 1 | (Bu4N)I | −7.12 | 0.85 |
| (Bu4N)Br | −8.54 | 1.48 | |
| (Bu4N)Cl | −9.75 | 1.96 | |
| 2 | (Bu4N)I | −2.23 | 1.11 |
| (Bu4N)Br | −5.85 | 2.14 | |
| (Bu4N)Cl | −9.56 | 2.84 | |
A final point of interest concerns the linearity of the chalcogen bonds and the geometrical relationship between the maximum of the MESP, the extension of the C–Se bond axis, and the Se⋯X− axis (Fig. 6; Table 4). In halogen-bonded systems, these three axes tend to coincide closely, whereas there is growing evidence in the literature24 for a misalignment of these in chalcogen-bonded systems. For the ChB donors reported here, we observe a deviation of VS,max from the extension of the C–Se bond axis of 16 to 18°; this increases to 21 to 26° in the cocrystals (see angle θ in Fig. 6). Furthermore, the Se⋯X− axis also deviates from the extension of the C–Se bond axis by 5 to 9° (φ in Fig. 6), but not always necessarily towards the presumably preferred binding site at VS,max. An inspection of the crystal structures suggests that this is a steric effect.
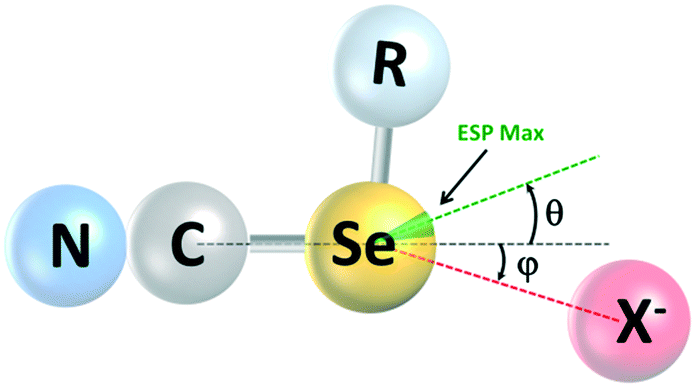 | ||
| Fig. 6 Local geometry of the ChB to selenium, indicating the angles between the extension of the C–Se bond axis and: (i) the maximum of the ESP (θ); (ii) the Se⋯X− axis (φ). VS,max and X− may or may not reside on the same side of the bond extension. | ||
Conclusions
In summary, we have demonstrated the strong aptitude of benzylic selenocyanates for the recognition of onium halides in the solid state and in solution. Single-crystal X-ray diffraction shows that the benzylic selenocyanate derivatives can adapt their conformations to bind halide anions. Anion binding activity has been established qualitatively in solution via13C and 77Se NMR spectroscopy. Taken together with other recent literature reports,16 one may speculate that benzylic selenocyanates may evolve into so-called ‘iconic’ ChB donors, in parallel with the iodoperfluorobenzenes which are widely used as iconic XB donors.Experimental
Synthesis of benzylic selenocyanate 1
A solution of potassium selenocyanate (0.8 g, 5.4 mmol, 2.7 eq.) in anhydrous DMF (15 mL) was added dropwise over a period of 30 min to a solution of α,α'-dibromo-p-xylene (0.5 g, 1.89 mmol, 1 eq.) under an argon atmosphere. Upon stirring, the solution gets cloudy. The reaction was monitored by thin layer chromatography (eluent petroleum ether/ether 1/2). After completion of the reaction (typically 45 min), addition of 60 mL of warm water (40 °C) precipitated an off-white solid. Further, the solid was washed twice with 50 mL of warm water (40 °C) in an ultrasonic bath for 5 min. The solid was filtered using Whatman filter paper and dried overnight at 80 °C. 96% yield, white solid.Melting point: 149–152 °C. 1H NMR (300 MHz, DMSO-d6) δ in ppm 7.37 (s, 4H), 4.31 (s, 4H). 13C NMR (75 MHz, DMSO-d6): 138.4 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 129.6 (C–H), 105.4 (CN), 32.77 (CH2). 77Se NMR (DMSO-d6): 321.2 (2Se, s).
C), 129.6 (C–H), 105.4 (CN), 32.77 (CH2). 77Se NMR (DMSO-d6): 321.2 (2Se, s).
Synthesis of benzylic selenocyanate 2
A solution of potassium selenocyanate (1.9 g, 13 mmol, 6 eq.) in anhydrous DMF (25 mL) is added dropwise over a period of 30 min to a solution of 1,2,4,5-tetrakis(bromomethyl) benzene (1 g, 2 mmol, 1 eq.) under an argon atmosphere. Upon addition, the solution turns pink and slowly turns cloudy. The reaction was monitored by thin layer chromatography (eluent petroleum ether/ether 1/2). After completion of the reaction (typically 30 to 40 min), addition of 60 mL of warm water (40 °C) precipitated an off-white solid. Further, the solid was washed twice with 60 mL of warm water (40 °C) in an ultrasonic bath for 5 min. The solid was filtered using Whatman filter paper and dried overnight at 80 °C. 95% yield, white solid.Melting point: 158–163 °C (decomposes). 1H NMR (300 MHz, DMSO-d6) δ in ppm 7.38 (s, 2H), 4.45 (s, 8H). 13C NMR (75 MHz, DMSO-d6): 136.83 (CC), 133.86 (C–H), 105.0 (CN), 29.6 (CH2). 77Se NMR (DMSO-d6): 311.12 (4Se, s).
Preparation of benzylic selenocyanate onium halide cocrystals
Benzene selenocyanates (1 and 2) and tetrabutyl ammonium halides (iodide, bromide, and chloride) were dissolved separately in acetone (2 equivalents of onium halides for ChB donor 1 and 4 equivalents of onium halides for ChB donor 2). The two solutions were mixed in a small borosilicate glass vial which was placed in a larger vial containing diethyl ether as a second less efficient solvent and the system was sealed. Ether was allowed to diffuse until crystals were formed. See also Table S.1† for additional conditions.Powder X-ray studies
Crystalline powdered pure benzene selenocyanates, onium halides, and cocrystals were individually packed in an aluminium sample holder and data sets were collected on a Rigaku Ultima IV powder diffractometer at 293 K (±2) (CuKα1 radiation with a wavelength of λ = 1.54056 Å). The measurements were carried out in focused beam geometry with a step-scan technique in 2θ range of 5–50°. Data were acquired by scintillation counter detector in continuous scanning mode with a step size of 0.02°.Single crystal X-ray studies
The crystals were mounted on glass fibres with glue prior to data collection. Crystals were cooled to 200 ± 2 K. The data were collected on a Bruker AXS diffractometer equipped with MoKα radiation (wavelength of λ = 0.7103 Å) with an APEX II CCD detector. The raw data collection and processing were performed with the Bruker APEX II software package. Structure solution and the refinement details are provided in ESI† S.3.Solution NMR studies
For 77Se, experimental setup and calibrations were performed using selenous acid (1 M solution in D2O); the chemical shift of the Se resonance was set to 1300.1 ppm. Exactly 0.25 M solutions of ChB donors (1 and 2) in DMSO-d6 were used to measure the 77Se and 13C NMR spectra. 13C NMR spectra were referenced using the solvent peak as a secondary reference. The amount of added ChB acceptor was chosen in order to form a solution containing equimolar amounts of ChB donor and ChB acceptor atoms (ESI† S.4).Conflicts of interest
There are no conflicts to declare.Acknowledgements
VK, CL, and DLB thank Dr. Bulat Gabidullin, Dr. Glenn Facey, and Dr. Peter Pallister for technical support and useful discussions. DLB thanks NSERC for funding. We are grateful to an anonymous reviewer for valuable feedback.References
-
(a)
G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, OUP, Oxford, 1999 Search PubMed
; (b) V. Kumar, T. Pilati, G. Terraneo, G. Ciancaleoni, A. Macchioni, G. Resnati and P. Metrangolo, Angew. Chem., 2018, 130, 1341 CrossRef
-
(a) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef PubMed
-
(a) D. J. Pascoe, K. B. Ling and S. L. Cockroft, J. Am. Chem. Soc., 2017, 139, 15160–15167 CrossRef PubMed
-
(a) Y. Zhang and W. Wang, Crystals, 2018, 8, 163, DOI:10.3390/cryst8040163
-
(a) S. Benz, J. Mareda, C. Besnard, N. Sakai and S. Matile, Chem. Sci., 2017, 8, 8164–8169 RSC
- L. Chen, J. Xiang, Y. Zhao and Q. Yan, J. Am. Chem. Soc., 2018, 140, 7079–7082 CrossRef PubMed
- S. P. Thomas, K. Satheeshkumar, G. Mugesh and T. N. Guru Row, Chem. – Eur. J., 2015, 21, 6793–6800 CrossRef PubMed
-
(a) A. F. Cozzolino, P. J. W. Elder and I. Vargas-Baca, Coord. Chem. Rev., 2011, 255, 1426–1438 CrossRef
- A. Mishra, C.-Q. Ma and P. Bäuerle, Chem. Rev., 2009, 109, 1141–1276 CrossRef PubMed
- P. C. Ho, P. Szydlowski, J. Sinclair, P. J. W. Elder, J. Kubel, C. Gendy, L. M. Lee, H. Jenkins, J. F. Britten, D. R. Morim and I. Vargas-Baca, Nat. Commun., 2016, 7, 11299–11309 CrossRef PubMed
- M. E. Brezgunova, J. Lieffrig, E. Aubert, S. Dahaoui, P. Fertey, S. Lebègue, J. Angyan, M. Fourmigué and E. Espinosa, Cryst. Growth Des., 2013, 13, 3283–3289 CrossRef
- S. Benz, J. López-Andarias, J. Mareda, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2017, 56, 812–815 CrossRef PubMed
- P. Wonner, L. Vogel, M. Düser, L. Gomes, F. Kniep, B. Mallick, D. B. Werz and S. M. Huber, Angew. Chem., Int. Ed., 2017, 56, 12009–12012 CrossRef PubMed
-
(a) K.-H. Linke and F. Lemmer, Z. Anorg. Allg. Chem., 1966, 345, 211–216 CrossRef
- A. Lari, R. Gleiter and F. Rominger, Eur. J. Org. Chem., 2009, 2267–2274 CrossRef
-
(a) H.-T. Huynh, O. Jeannin and M. Fourmigué, Chem. Commun., 2017, 53, 8467–8469 RSC
-
(a) G. E. Garrett, G. L. Gibson, R. N. Straus, D. S. Seferos and M. S. Taylor, J. Am. Chem. Soc., 2015, 137, 4126–4133 CrossRef PubMed
- M. Hojjatie, S. Muralidharan and H. Freiser, Tetrahedron, 1989, 45, 1611–1622 CrossRef
- S. Scheiner, Chem. – Eur. J., 2016, 22, 18850–18858 CrossRef PubMed
-
(a) P. A. Gale and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3595–3596 RSC
-
(a) N. G. White and M. J. MacLachlan, Chem. Sci., 2015, 6, 6245–6249 RSC
-
(a) V. Kumar, T. Pilati, G. Terraneo, F. Meyer, P. Metrangolo and G. Resnati, Chem. Sci., 2017, 8, 1801–1810 RSC
-
(a) M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC
- P. Politzer, J. S. Murray, T. Clark and G. Resnati, Phys. Chem. Chem. Phys., 2017, 19, 32166–32178 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic and NMR data. CCDC: 1849844–1849846 and 1851129. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ce01365a |
| This journal is © The Royal Society of Chemistry 2018 |