
Co-crystallization of 1,3,5-trifluoro-2,4,6-triiodobenzene (1,3,5-TFTIB) with a variety of Lewis bases through halogen-bonding interactions
Xue-Hua
Ding†
 a,
Chang-Jin
Ou†
ab,
Shi
Wang
b,
Ling-Hai
Xie
b,
Jin-Yi
Lin
a,
Jian-Pu
Wang
*a and
Wei
Huang
*abc
a,
Chang-Jin
Ou†
ab,
Shi
Wang
b,
Ling-Hai
Xie
b,
Jin-Yi
Lin
a,
Jian-Pu
Wang
*a and
Wei
Huang
*abc
aKey Laboratory of Flexible Electronics (KLOFE) & Institute of Advanced Materials (IAM), Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), Nanjing Tech University (NanjingTech), 30 South Puzhu Road, Nanjing 211816, China. E-mail: iamjpwang@njtech.edu.cn; iamwhuang@njtech.edu.cn
bKey Laboratory for Organic Electronics & Information Displays (KLOEID) and Institute of Advanced Materials (IAM), Jiangsu National Synergistic Innovation Center for Advanced Materials (SICAM), Nanjing University of Posts & Telecommunications (NUPT), Nanjing 210023, China
cShaanxi Institute of Flexible Electronics (SIFE), Northwestern Polytechnical University (NPU), 127 West Youyi Road, Xi'an 710072, Shaanxi, China
First published on 18th August 2017
Abstract
The halogen bond is emerging as an important driving force for supramolecular self-assembly, and has attracted great interest in the past decade. Among the ample halogen-bonding donors, we note the ability of 1,3,5-trifluoro-2,4,6-triiodobenzene (1,3,5-TFTIB) to co-crystallize with various halogen-bonding acceptors, ranging from neutral Lewis bases (nitrogen-containing heterocycles, N-oxides, and triphenylphosphine selenide (Ph3PSe)) to anions (halide ions and thiocyanate ion), leading to a wide diversity of supramolecular architectures. Some of them are promising optoelectronic functional materials. In this review, we concentrate on the structures of multicomponent supramolecular complexes, highlight typical examples, and point out the main possible directions that remain to be developed in this field. From the perspectives of supramolecular chemistry and crystal engineering, the complexes reviewed here should provide useful information for further design and investigation on this fascinating class of halogen-bonding supramolecular architectures.
1. Introduction
Since Jean-Marie Lehn's famous description of supramolecular chemistry, supramolecular synthesis as a rapidly growing field is still in its formative stages.1–5 The co-crystallization strategy in crystal engineering offers a rational approach to the design and synthesis of multi-component supramolecular complexes (such as co-crystals and salts) with desired structures and special functionalities,6–12 which gain an advantage over single-component compounds on the control of molecular arrangements and physicochemical properties.13 The co-crystallization process has been largely associated with molecular recognition and self-assembly between components driven by non-covalent interactions. Hence, the understanding of non-covalent interactions is of great importance, for instance, hydrogen bonds, halogen bonds, π–π stacking, van der Waals forces, etc.14–16Currently, focus has been extended towards halogen bonds from the better-known hydrogen bond since these have been proven to be an alternative powerful tool in supramolecular chemistry and crystal engineering.17–21 The relevant research on halogen bond covers a wide scope from fundamental studies (for example, the nature of a halogen bond22–26 and the construction of novel supramolecular architectures27,28) and materials sciences (optoelectronic materials,29,30 anion recognition,31–34 liquid crystals,35 capture and release of small molecules,36 supramolecular gels,37 and so forth) to biological systems.38,39
Recent developments have significantly enhanced our understanding of halogen-bonding interactions. A halogen bond (R–X⋯Y) occurs when an attractive interaction exists between the electrophilic region on a halogen atom (the so-called σ-hole) and an electron donor (Lewis base), sharing many similar features with a hydrogen bond.40–42 The electron-deficient halogen atom (X = Cl, Br, I, named the halogen-bonding donor) acts as an electron acceptor of the electron-rich Lewis base (Y = N, O, S, P or halogen atoms, called the halogen-bonding acceptor). The distance (X⋯Y) between the halogen atom and the electron donor atom is shorter than the sum of the van der Waals radii, and the corresponding bond angle (R–X⋯Y) is close to 180°.41 In addition to the conventional linear halogen bond, two types of halogen⋯halogen contacts (R–X1⋯X2–Z) are frequently encountered in the crystal structures of halogen-substituted molecules, where contacts with θ1 (R–X1⋯X2) ≈ θ2 (X1⋯X2–Z) were defined as type I, and type II with θ1 ≈ 180° and θ2 ≈ 90°.43
The halogen-bonding strength increases in the order of Cl<Br<I depending on their electronegativity,44 and can be further enhanced by introducing electron-withdrawing groups to the halogen-bonding donor, such as the fluorine atom.45,46 For this reason, perfluorinated 1,3,5-trifluoro-2,4,6-triiodobenzene (1,3,5-TFTIB) was envisaged as an ideal halogen-bonding donor and has been widely used for the construction of various supramolecular architectures by co-crystallizing with diverse halogen-bonding acceptors, ranging from neutral Lewis bases (nitrogen-containing heterocycles, N-oxides, and triphenylphosphine selenide (Ph3PSe)) to anions (halide ions and the thiocyanate ion) (Scheme 1). In view of its potential in the design and synthesis of new multicomponent crystalline materials, we believe that it is timely and necessary to give an overview of the research progress on 1,3,5-TFTIB. In this review, our attention is focused on the structures of multicomponent supramolecular complexes, which were investigated by crystallographic and theoretical methods.
 | ||
| Scheme 1 Chemical structures of most Lewis bases in order of their appearance. | ||
2. Co-crystallization of 1,3,5-TFTIB with various halogen-bonding acceptors
2.1. Nitrogen-containing heterocycles as the halogen-bonding acceptors
2.1.1.1. Pyridine derivatives. The co-crystals concerning 1,3,5-TFTIB were first reported by van der Boom and coworkers, with bipyridyl derivatives (bis-1,4-(4-pyridylethyl-enyl)-benzene (BPEB), trans-1,2-bis(4-pyridyl)-ethylene (BPE) and 4,4′-bipyridine (BPY)) as halogen-bonding acceptors.47 In all three cases, the halogen-bonding donor and acceptor are inclined to co-crystallize in a 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio instead of the anticipated 2:3 ratio, and only two iodine atoms in 1,3,5-TFTIB are involved in halogen bonds rather than 3-fold C–I⋯N intermolecular interactions. Intermolecular C–I⋯N halogen bonds, in combination with the π-stacking between aromatic systems and C–I⋯F contacts, afford interesting supramolecular architectures. There seems to be no apparent correlation between the size/length of bipyridyl derivatives and the distance of these intermolecular interactions. DFT calculations were performed to investigate the nature of the interaction between the pyridine nitrogen and 1,3,5-TFTIB, and the results demonstrated a weakening of N⋯I interactions as more electron-donor moieties coordinate to 1,3,5-TFTIB, which may be a contributing factor to the experimental findings that the number of halogen bonds on a single aromatic halogen-bonding donor is limited. Alternatively, another likely reason was postulated to be the favorable packing arrangements.
:1 molar ratio instead of the anticipated 2:3 ratio, and only two iodine atoms in 1,3,5-TFTIB are involved in halogen bonds rather than 3-fold C–I⋯N intermolecular interactions. Intermolecular C–I⋯N halogen bonds, in combination with the π-stacking between aromatic systems and C–I⋯F contacts, afford interesting supramolecular architectures. There seems to be no apparent correlation between the size/length of bipyridyl derivatives and the distance of these intermolecular interactions. DFT calculations were performed to investigate the nature of the interaction between the pyridine nitrogen and 1,3,5-TFTIB, and the results demonstrated a weakening of N⋯I interactions as more electron-donor moieties coordinate to 1,3,5-TFTIB, which may be a contributing factor to the experimental findings that the number of halogen bonds on a single aromatic halogen-bonding donor is limited. Alternatively, another likely reason was postulated to be the favorable packing arrangements.
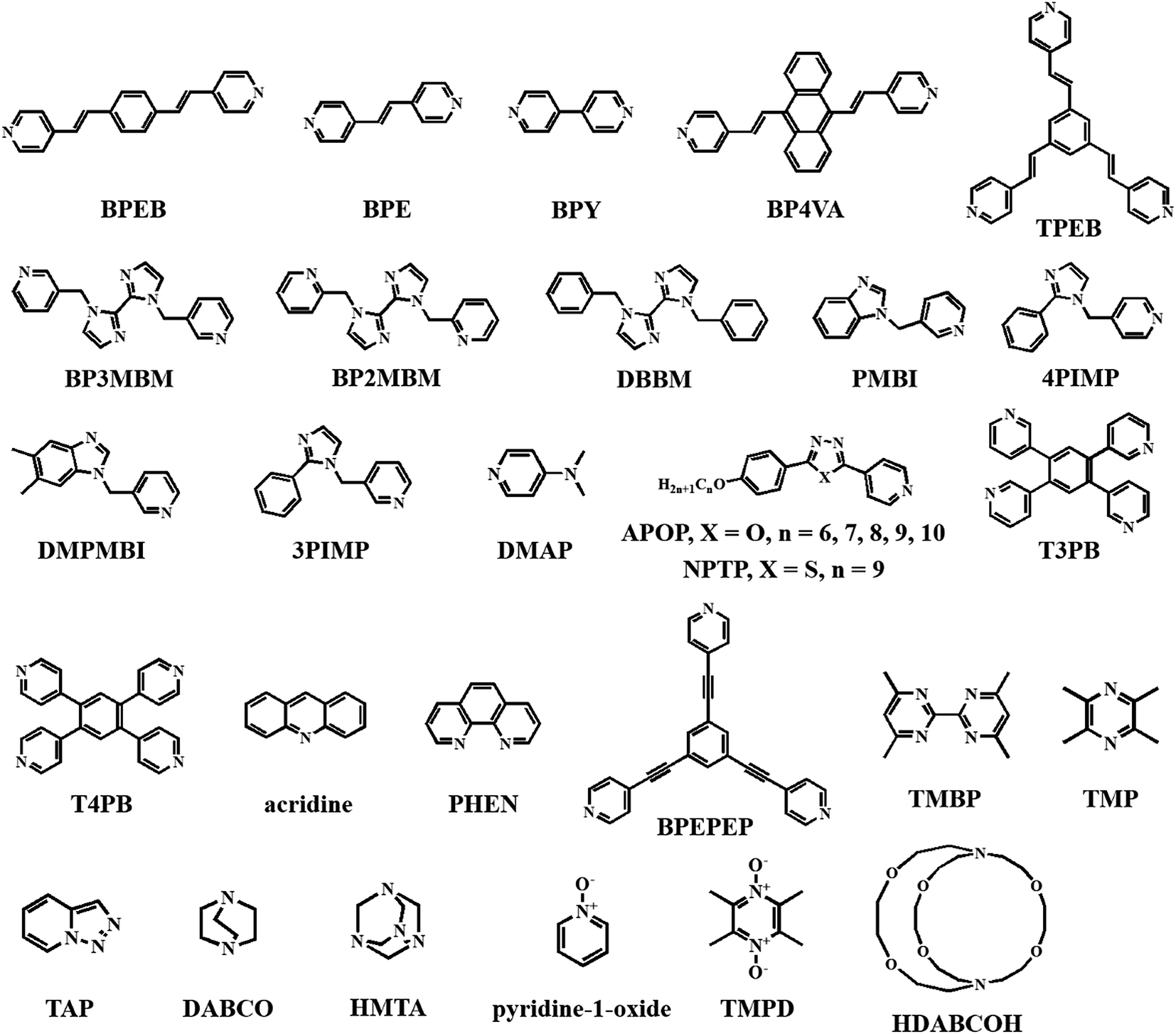
Recently, a comment was made on the interpenetrated network structure of 1,3,5-TFTIB-BPY by Schöellhorn et al.,48 while 1,3,5-TFTIB-BPE was synthetized at the micro-/nanoscale by Hu's group to gain insight into the “assembly–structure–charge transfer (CT)–properties” relationship.49 In the latter case, wire-like crystals with segregated stacking are measured to be quasi-1D semiconductors and exhibit strong violet-blue photoluminescence (PL) from the lowest CT1 excitons (ΦPL = 26.1%), which can be confined and propagate oppositely along the 1D axial direction (Fig. 1). Furthermore, the appearance of CT interactions has been confirmed by relevant experiments and DFT calculations, and is attributed to the π electron-rich character in the BPE columns, which promotes the CT process from the donor BPE to the acceptor 1,3,5-TFTIB. Impressively, it gives a deep understanding of the relationship among molecular packing, CT interactions and optoelectronic properties in halogen-bonding co-crystals.
 | ||
| Fig. 1 (a) Intermolecular interactions and molecular packing structures of 1,3,5-TFTIB-BPE. (b) Schematic diagrams of segregated stacking. (c) Confocal laser scanning microscopy (CLSM) images. (d) Electric conductivity. Reprinted with permission from ref. 49. Copyright 2015 The American Chemical Society. | ||
A larger conjugated bipyridyl acceptor, 9,10-bis((E)-2-(pyridin-4-yl)vinyl)anthracene (BP4VA), was investigated later, which is a typical aggregation-induced emission (AIE) luminogen.50 Two polymorphs of co-crystals C1 and C2 were prepared by the self-assembly of BP4VA with 1,3,5-TFTIB in a 1:1 molar ratio, a THF solution for C1 while a THF and ethanol mixed solvent for C2. The two polymorphs crystallized in the monoclinic space group P21/c and triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) respectively, in which BP4VA is highly twisted with dihedral angles of 66.09° for C1 and 54.32° for C2 between the central anthracene and the double bond (Fig. 2a and d). Although both packed in segregated-stacking modes, the block-like C1 displays strong H-type interactions between BP4VA and 1,3,5-TFTIB molecules due to strong π⋯π interactions, whereas for needle-like C2, the BP4VA molecules adopt J-aggregation (Fig. 2b, c, e and f). In contrast with the pure BP4VA crystal, the formation of H-aggregates in C1 probably leads to non-radiative decay in the highly interacting solid state, and there are no obvious π-interactions in C2, which may allow efficient radiative transition and enhanced fluorescence emission. As was expected, C1 shows a weak green emission peaking at 510 nm with a low quantum yield ϕF of 2%, but C2 exhibits a bright yellow emission located at 554 nm with a high ϕF of 34%. In addition, a unique mechanochromic behavior was observed in C1.
respectively, in which BP4VA is highly twisted with dihedral angles of 66.09° for C1 and 54.32° for C2 between the central anthracene and the double bond (Fig. 2a and d). Although both packed in segregated-stacking modes, the block-like C1 displays strong H-type interactions between BP4VA and 1,3,5-TFTIB molecules due to strong π⋯π interactions, whereas for needle-like C2, the BP4VA molecules adopt J-aggregation (Fig. 2b, c, e and f). In contrast with the pure BP4VA crystal, the formation of H-aggregates in C1 probably leads to non-radiative decay in the highly interacting solid state, and there are no obvious π-interactions in C2, which may allow efficient radiative transition and enhanced fluorescence emission. As was expected, C1 shows a weak green emission peaking at 510 nm with a low quantum yield ϕF of 2%, but C2 exhibits a bright yellow emission located at 554 nm with a high ϕF of 34%. In addition, a unique mechanochromic behavior was observed in C1.
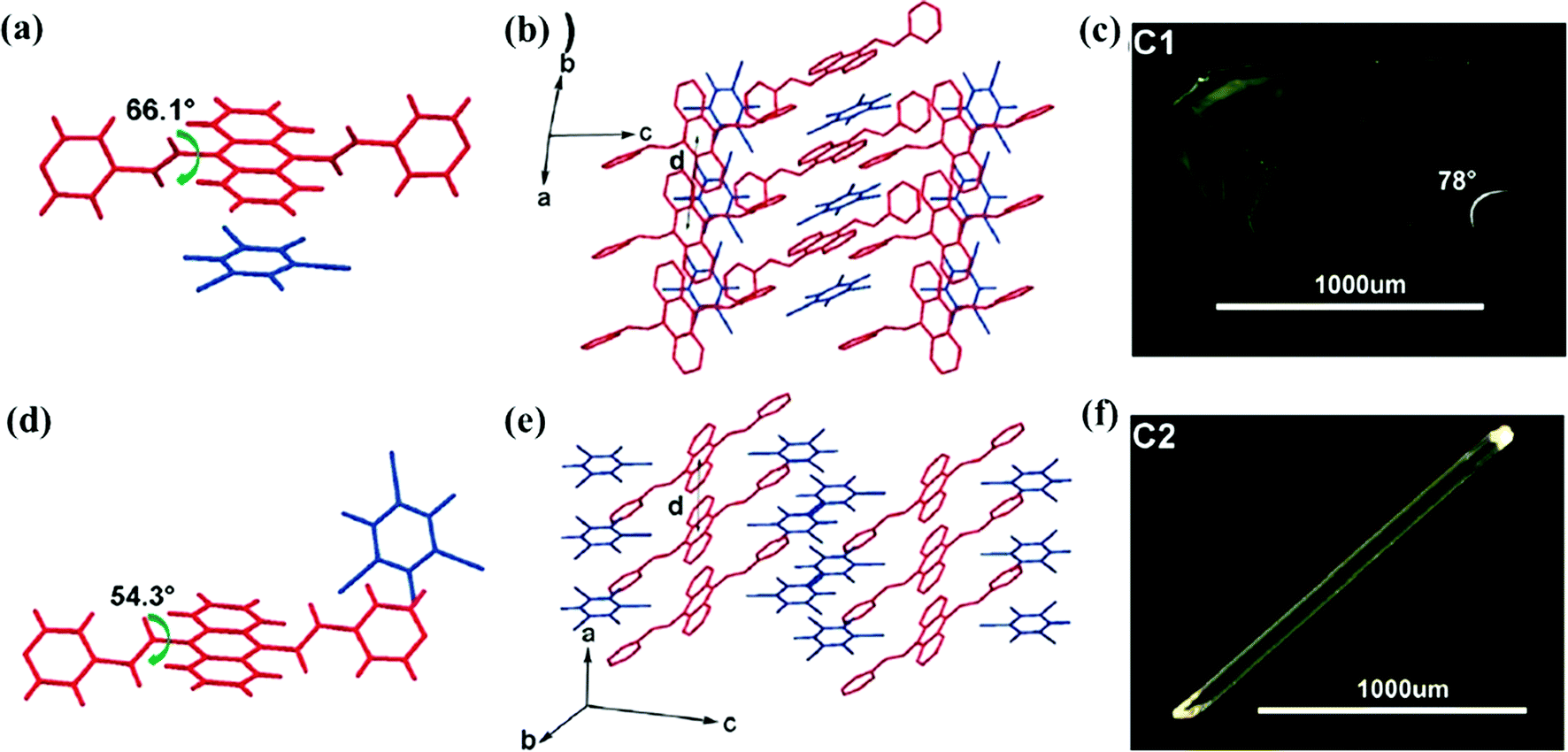 | ||
| Fig. 2 Two polymorphs C1 and C2 of 1,3,5-TFTIB-BP4VA. Conformational structure (a), stacking of the molecular columns (b) and optical image (c) of C1. Conformational structure (d), stacking of the molecular columns (e) and optical image (f) of C2. Reprinted with permission from ref. 50. Copyright 2017 The Royal Society of Chemistry. | ||
When extending this work to the tripyridyl system, a 1:2 co-crystal was obtained by slow evaporation of the CHCl3 solution containing 1,3,5-tris[4-pyridyl(ethenyl)]benzene (TPEB) and 1,3,5-TFTIB.51 X-ray crystallographic analysis revealed the formation of a halogen-bonding architecture, with both tripyridyl component and 1,3,5-TFTIB involved in π–π stacking interactions. Two nitrogen atoms from TPEB feature a strong and single C–I⋯N halogen bond (dN⋯I = 2.812 Å, ∠C–I⋯N = 176.59°) with two 1,3,5-TFTIB molecules, and the third nitrogen atom forms two relatively weak C–I⋯N halogen bonds (dN⋯I = 3.244 Å, ∠C–I⋯N = 158.87°) with the other two 1,3,5-TFTIB molecules (Fig. 3a). After two N atoms transfer their electron density to 1,3,5-TFTIB, the lower electron density in the third N atom may be responsible for the formation of a different halogen bond. As described in Fig. 3b, tripyridyl molecules are aligned in parallel, whereas 1,3,5-TFTIB molecules consist of two types of planes with a 41.7° angle.
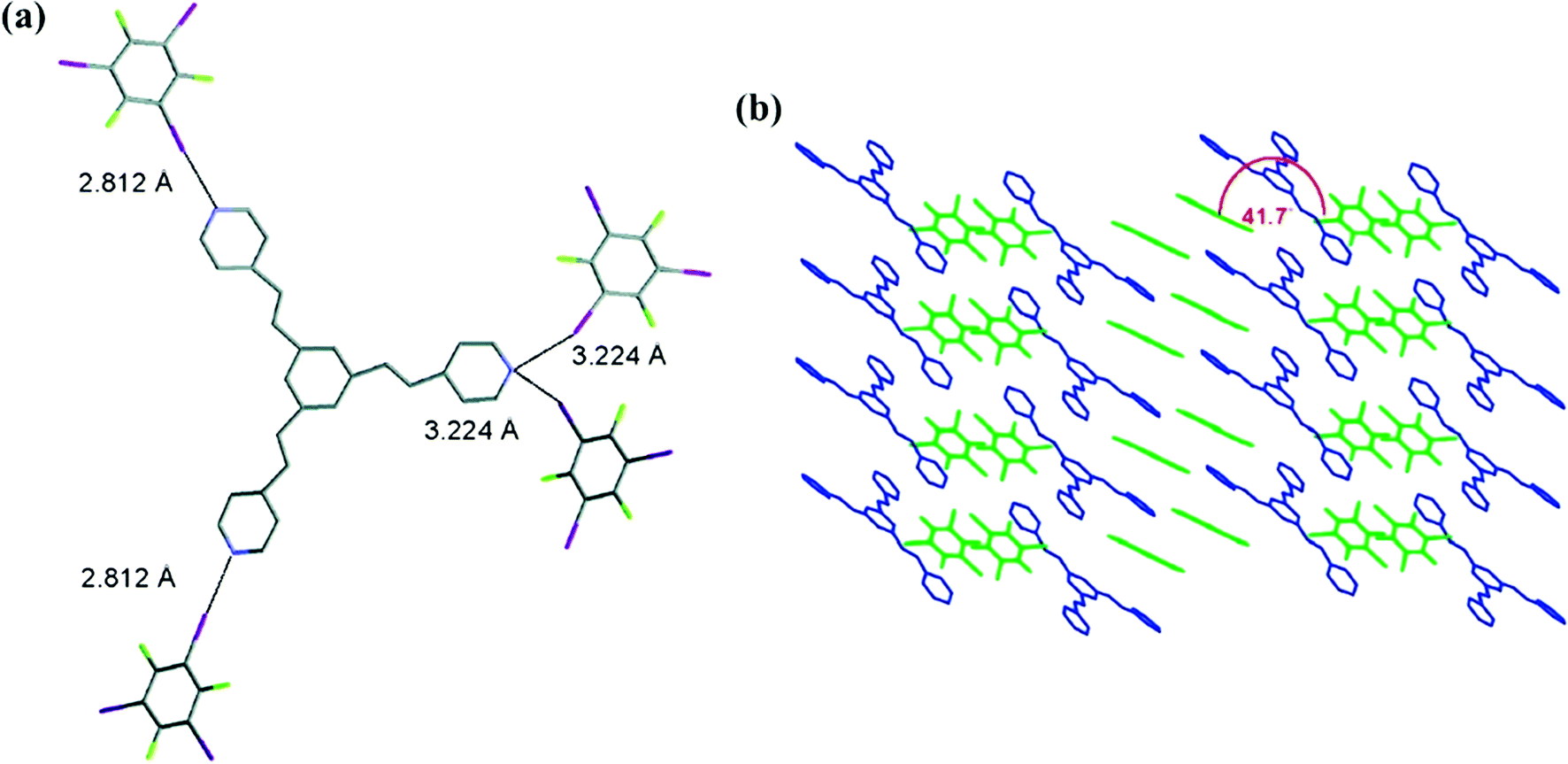 | ||
| Fig. 3 (a) TPEB is halogen bonded to four 1,3,5-TFTIB molecules through C–I⋯N interactions. (b) Schematic diagrams of crystal packing. Reprinted with permission from ref. 51. Copyright 2008 The American Chemical Society. | ||
In a general way, halogen bonds follow the best-donor/best-acceptor rules like hydrogen bonds and the “better” halogen-bonding donor preferentially interacts with the acceptor atom with a larger negative electrostatic potential. With a view to establishing how effectively the ranking and hierarchy of acceptor sites based on molecular electrostatic potential translate into predictable primary interactions, Aakeröy et al. conducted systematic co-crystallizations on three multi-topic N-heterocyclic acceptors, namely 1,1′-bis(pyridin-3-ylmethyl)-2,2′-biimidazole (BP3MBM), 1,1′-bis(pyridin-2-ylmethyl)-2,2′-biimidazole (BP2MBM) and 1,1′-dibenzyl-2,2′-biimidazole (DBBM).52 Thereinto, the latter one is employed as a control experiment to ensure that observed binding preferences are not simply the result of steric hindrance. Electrostatic potential calculations state clearly that pyridine nitrogen atoms carry the more negative electrostatic potential than those in the imidazole making them the better acceptors.
Indeed, theoretical calculations are consistent with the experimental results. In the co-crystal between 1,3,5-TFTIB and BP3MBM, both pyridine and imidazole N atoms participate in the halogen-bonding interactions (Fig. 4a), and the C–I⋯N(im) distances are much longer than the C–I⋯N(py) length, giving an indication that N(py) is the preferred binding site. Besides, a combination of intermolecular C–I⋯F halogen bonds and C–H⋯N(im) hydrogen bonds together drive the formation of supramolecular architectures.
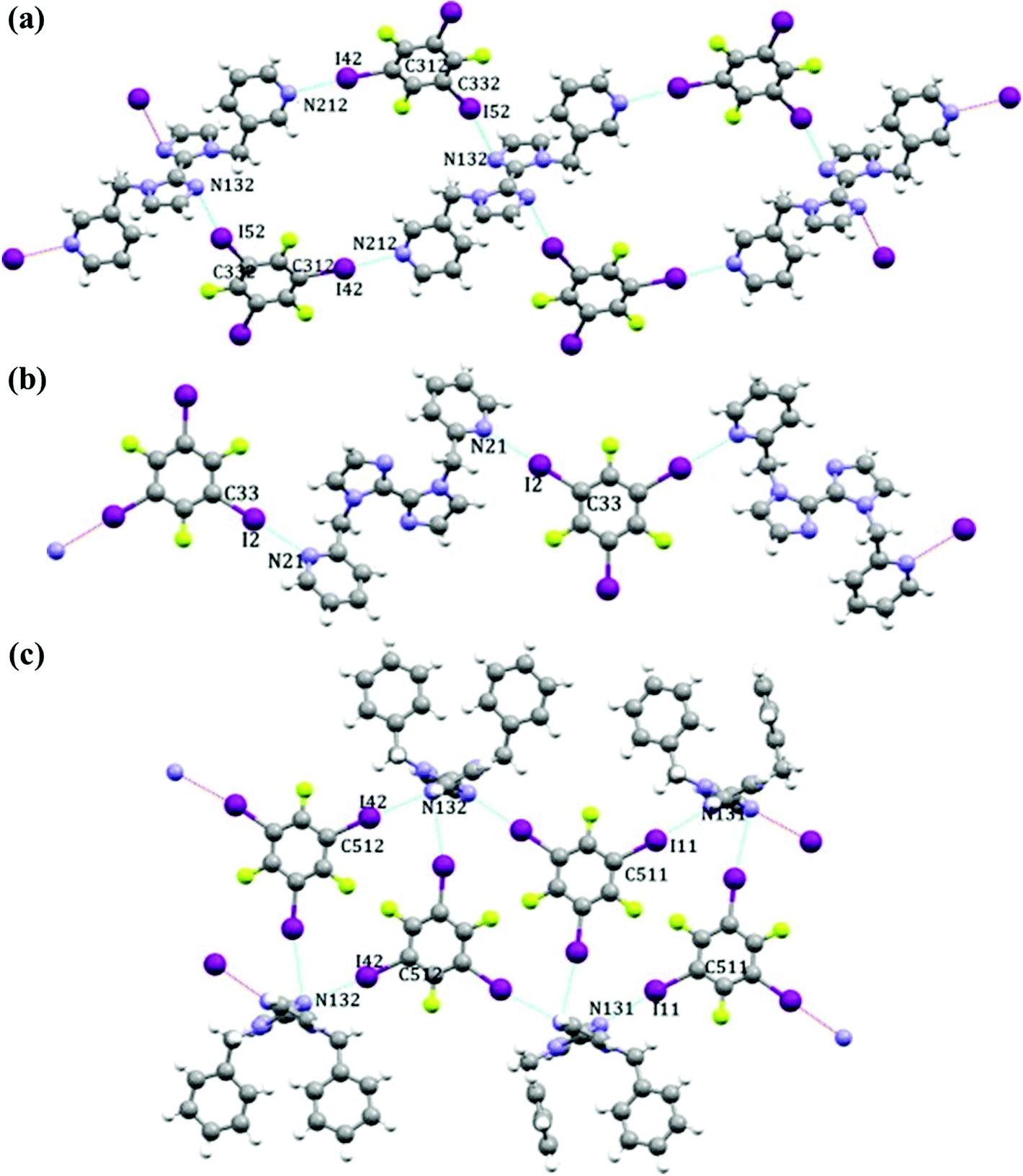 | ||
| Fig. 4 Primary halogen-bonding interactions in 1,3,5-TFTIB-BP3MBM (a), 1,3,5-TFTIB-BP2MBM (b) and 1,3,5-TFTIB-DBBM (c). Reprinted with permission from ref. 52. Copyright 2014 Elsevier. | ||
As for the system of 1,3,5-TFTIB and BP2MBM, two iodine atoms are halogen bonded to pyridine nitrogen atoms forming the 1D zig-zag chains without imidazole nitrogen atoms involved in any significant short contacts (Fig. 4b). In terms of the control group, terminal phenyl groups as ‘arms’ of the acceptor are unexpected to appear on the same side of the aromatic core. One of the N(im) atoms is engaged in a near-linear halogen bond while the other one generates dissymmetric bifurcated halogen bonds with two adjacent 1,3,5-TFTIB molecules (Fig. 4c). Such bifurcated halogen bonds are rare, as nitrogen atom only has one lone pair and tends to form one non-covalent interaction with a single electron-pair acceptor.
Subsequently, several pyridine-containing imidazoles with two different acceptor sites were utilized to react with 1,3,5-TFTIB, i.e. 1-(pyridin-3-ylmethyl)-benzimidazole (PMBI), 4-((2-phenyl-imidazol-1-yl)methyl)pyridine (4PIMP), 5,6-dimethyl-1-(pyridin-3-ylmethyl)-benzoimidazole (DMPMBI) and 3-((2-phenylimidazol-1-yl)methyl)pyridine (3PIMP).53 The “better” halogen-bonding donor shows a preference for the acceptor atom with a more negative electrostatic potential value, but the selectivity for binding sites is crucially dependent on the magnitude of potential-energy differences. If the difference in molecular electrostatic potential is not large enough, structural selectivity will be lost and both potential acceptor sites are simultaneously engaged in halogen bonds. As with every situation here, both N(py) and N(im) are halogen bonded to 1,3,5-TFTIB through C–I⋯N interactions. Furthermore, 4-dimethylaminopyridine (DMAP) was included in their study that has been previously reported by Bruce and co-workers.54 The difference in electrostatic potential reaches up to 167 kJ mol−1 and only the best acceptor site is preferred by the halogen-bonding donor, whereas the second-best acceptor is not involved in any short C–I⋯N contacts.
An extension of this work was carried out based on a series of 4-[5-(4-alkoxyphenyl)-1,3,4-oxadiazole-2-yl]pyridines with hexyloxy- to decyloxy- alkyl chains (APOP).55 In spite of the reactions from a 3:1 mixture of pyridine-containing oxadiazoles and 1,3,5-TFTIB, all complexes show a 1:1 stoichiometry and similar crystal packing, in which three nitrogen atoms and iodine atoms are all active in the formation of halogen bonds. A couple of 1,3,5-TFTIB connect each other through F⋯F interactions with a separation from 2.54 to 2.62 Å, and are linked to four oxadiazole molecules via C–I⋯N halogen-bonding interactions (Fig. 5a). The iodine atom para to this fluorine forms a halogen bond with the pyridine N atom of one oxadiazole molecule while the two ortho ones bind to the oxadiazole N atoms from a different molecule. The C–I⋯N distances do not change much with the alkyl chain length, in good agreement with the electronic structure calculations. The most negative electrostatic potential is on the oxadiazole nitrogen closest to the phenyl ring rather than the pyridine N atom that has the least negative value in all three nitrogen atoms unexpectedly. That is to say, the most favourable binding site would be the oxadiazole N atom nearest to the phenyl ring if the interaction with 1,3,5-TFTIB is only electrostatic in nature and no crystal packing effects are considered. However, the differences between the three nitrogen sites are small enough that all the N atoms simultaneously are involved in the formation of halogen bonds, and the C–I⋯N(ox) interaction is as strong as the classic C–I⋯N(py) interaction.
 | ||
| Fig. 5 (a) A couple of 1,3,5-TFTIB are halogen bonded to four 4-[5-(4-hexyloxyphenyl)-1,3,4-oxadiazole-2-yl]pyridine molecules. (b) Crystal packing in the co-crystal between 1,3,5-TFTIB and 4-[5-(4-nonyloxyphenyl)-1,3,4-thiadiazole-2-yl]pyridine, showing two adjacent layers (above in bold, below in light colour). Reprinted with permission from ref. 55. Copyright 2016 The Royal Society of Chemistry. | ||
The substitution of sulfur for the oxygen atom on the oxadiazole ring gives rise to a different supramolecular packing. For instance, the thiadiazole derivative, 4-[5-(4-nonyloxyphenyl)-1,3,4-thiadiazole-2-yl]pyridine (NPTP), when reacted with 1,3,5-TFTIB, produced a 2:1 co-crystal.55 In this case, only the pyridine nitrogen atom is favored, which can be rationalized by the change in the molecular electrostatic potential. Two iodine atoms from one 1,3,5-TFTIB molecule are linked with two thiadiazole molecules, giving rise to the V-shaped trimer that is staggered with its neighbours (Fig. 5b). It can be drawn from these results that the archetypal iodine⋯pyridine interaction is not necessarily the most favored when halogen-bonding acceptors such as oxadiazole are present. However, one can tune their electrostatic potential by using the appropriate strategy.
In order to explore the structural competition between π⋯π interactions and halogen bonds, two co-crystals have been prepared by Ji's group with 1,2,4,5-tetra(3-pyridyl)benzene (T3 PB) and 1,2,4,5-tetra(4-pyridyl)benzene (T4 PB) as the halogen-bonding acceptors.56 X-ray crystallographic analyses reveal that the π⋯π interactions between two 1,3,5-TFTIB molecules can successfully compete with intermolecular C–I⋯N halogen bonds. Quantum chemical calculations at the SCS-MP2/SDD** level of theory explain the structural competition well and confirm that the π⋯π interaction is much stronger than the C–I⋯N halogen bond, indicating that π⋯π interactions serve as the primary force in the co-crystallization process.
Self-assembly of 1,3,5-TFTIB with acridine or 1,10-phenanthroline (PHEN) has resulted in the formation of a halogen-bonding co-crystal with a 1: 1 molar ratio, both crystallizing in the P21/c space group.57 In the two co-crystals, the three iodine atoms all participate in C–I⋯N and C–I⋯I halogen bonds, which offer novel supramolecular frameworks. 13C and 19F solid-state magic-angle spinning (MAS) NMR is an effective tool to probe the formation of a halogen bond. To be specific, 13C solid-state NMR using 19F → 13C cross polarization (CP) from the halogen-bonding donor to the 13C nuclei of the acceptor indicates that the chemical shifts of the carbons bonded to iodine atoms increase in terms of the donor and gives a direct indication of co-crystallization. Upon the occurrence of a halogen bond, the 19F chemical shift decreases in the 19F solid-state NMR. Gauge-including projector-augmented wave density functional theory (GIPAW DFT) calculations of magnetic shielding constants provide final structures in best agreement with the experimental 13C and 19F chemical shifts.
Halogen bonding can also be applied in surface molecular engineering. The first 2D open porous halogen-bonding network on a highly oriented pyrolytic graphite (HOPG) surface has been fabricated by Wan's group, using the tripyridyl derivative (4-(2-(3,5-bis(2-(pyridin-4-yl)ethynyl)phenyl)ethynyl)pyridine (BPEPEP)) and 1,3,5-TFTIB.58 The electrical stimuli of a scanning tunneling microscopy (STM) tip can induce the formation of a binary supramolecular structure through halogen-bonding interactions. The proposed structural model and high-resolution STM image for the honeycomb network are given as depicted in Fig. 6. The lattice parameters were measured to be a = b = 2.5 ± 0.2 nm, γ = 60 ± 2°. The size of a typical honeycomb pore is in accordance with the expected size of 2.5 nm predicted by DFT calculations.
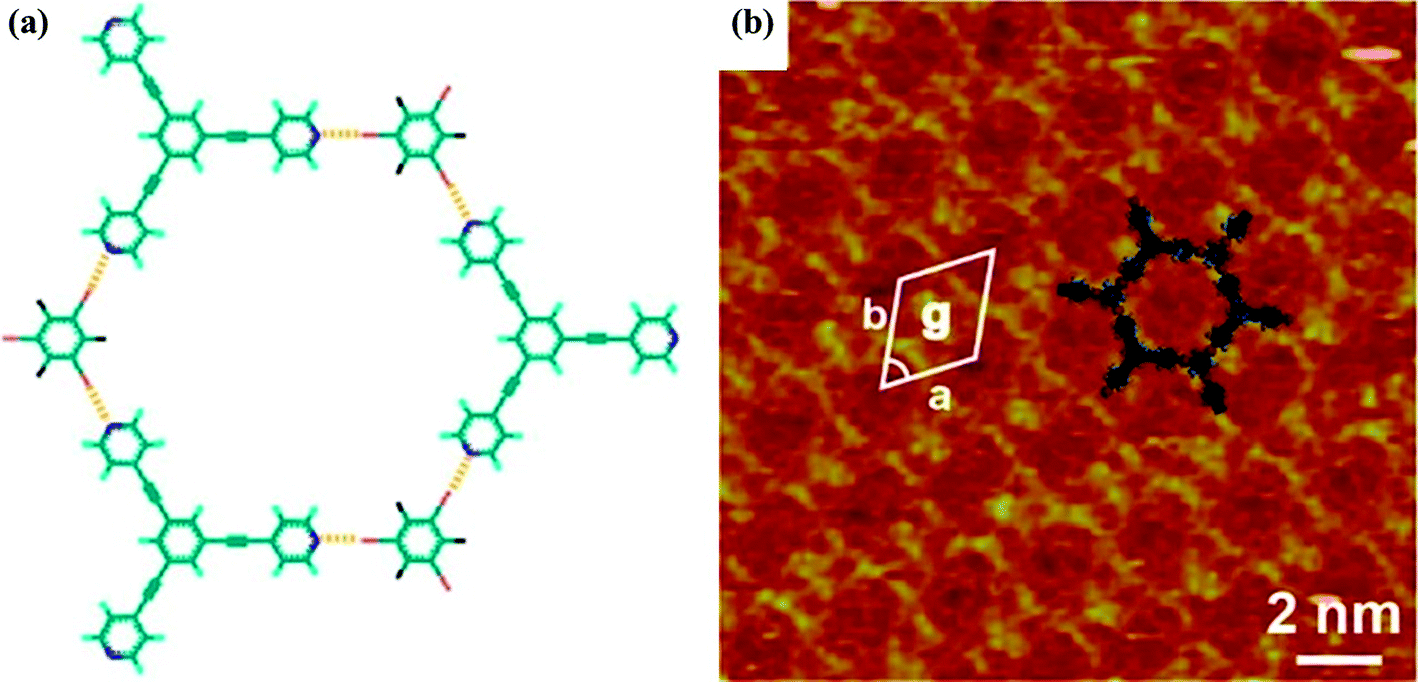 | ||
| Fig. 6 (a) Structural model of the honeycomb structure between 1,3,5-TFTIB and BPEPEP. Possible halogen bonds are depicted by yellow dashed lines. (b) High-resolution scanning tunneling microscopy (STM) image. Tunneling conditions: Vbias = 888 mV, It = 1060 pA. Reprinted with permission from ref. 58. Copyright 2015 The American Chemical Society. | ||
2.1.1.2. Other aromatic nitrogen-containing heterocycles. Apart from pyridine derivatives, other aromatic nitrogen-containing heterocycles can also act as halogen-bonding acceptors. In the case of 4,4′,6,6′-tetramethyl-2,2′-bipyrimidine (TMBP) co-crystallizing with 1,3,5-TFTIB, it was found that the π⋯π interactions between two 1,3,5-TFTIB molecules can successfully compete with intermolecular C–I⋯N halogen bonds by single-crystal X-ray diffraction analysis and quantum chemical calculations at the SCS-MP2/SDD** level of theory.56
2,3,5,6-Tetramethylpyrazine (TMP) was also chosen as the halogen-bonding acceptor to interact with 1,3,5-TFTIB, giving rise to a novel co-crystal that features two linear C–I⋯N halogen bonds, with angles of 178.09 and 179.81°, and lengths of 2.991 and 2.993 Å, respectively.57 As illustrated in Fig. 7, a zig-zag halogen-bonding chain is produced and linked with the adjacent one by C–I⋯F close contacts (dI⋯F = 3.133 Å), presenting 2D supramolecular networks. 13C and 19F solid-state NMR has been used to characterize structural features of the halogen-bonding donor and acceptor, along with GIPAW DFT calculations.
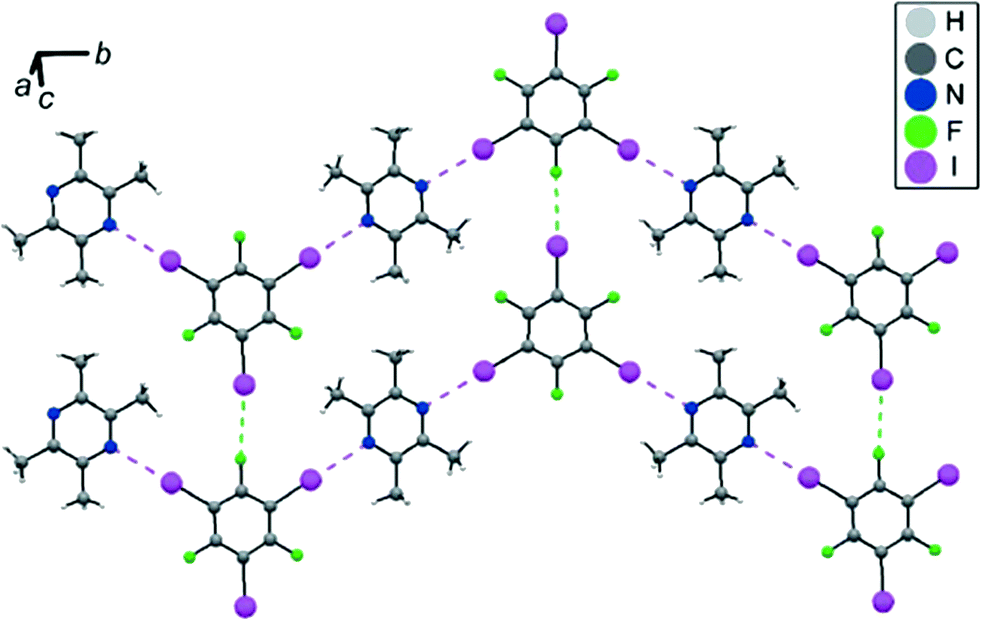 | ||
| Fig. 7 Crystal packing in 1,3,5-TFTIB-TMP. Reprinted with permission from ref. 57. Copyright 2017 The International Union of Crystallography. | ||
As described above, the selectivity for halogen-bonding binding sites is crucially dependent on the magnitude of potential-energy differences. When the difference in the molecular electrostatic potential is not large enough, as in the case for [1,2,3]triazolo-[3,5-α]pyridine (TAP), both potential acceptor sites are simultaneously involved in halogen bonds with 1,3,5-TFTIB through C–I⋯N interactions (Fig. 8) and structural selectivity is lost.59 DFT calculations on the triazole molecule reveal that the two possible acceptor sites have electrostatic potential values of −180 kJ mol−1 and −206 kJ mol−1, respectively, giving rise to a small difference (26 kJ mol−1).
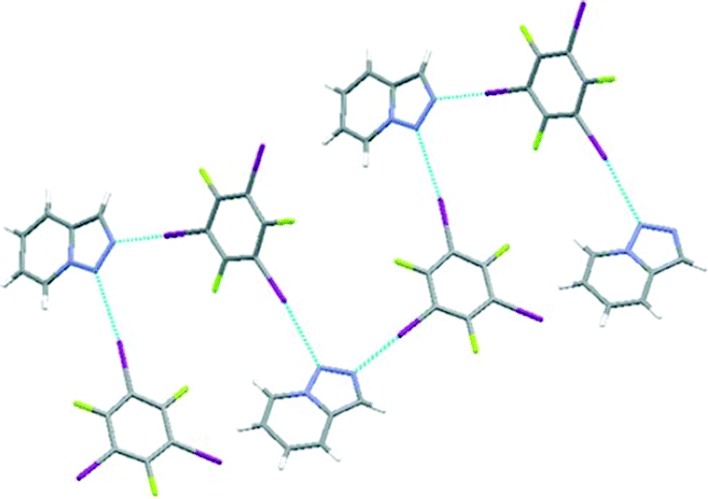 | ||
| Fig. 8 Primary halogen-bond interactions in 1,3,5-TFTIB-TAP. Reprinted with permission from ref. 59. Copyright 2016 The Royal Society of Chemistry. | ||
:acetone:water = 2:7:7), even though the initial crystallization experiment was carried out in a 3:2 stoichiometric ratio of DABCO:1,3,5-TFTIB.60 In the co-crystal, DABCO was halogen bonded to two 1,3,5-TFTIB molecules through C–I⋯N interactions. However, 1,3,5-TFTIB can be described as “unsaturated” in a supramolecular sense, where one of iodine atoms is involved in two halogen bonds (as both donor and acceptor), another is a halogen-bonding acceptor, and the third one does not participate in a halogen-bonding interaction. The I⋯I halogen bonds connect adjacent trimers into 2D tapes that propagate parallel to the ab plane (Fig. 9a). The π-stacking interactions between 1,3,5-TFTIB molecules from adjacent 2D tapes lead to their interdigitation (but not interpenetration) (Fig. 9b).
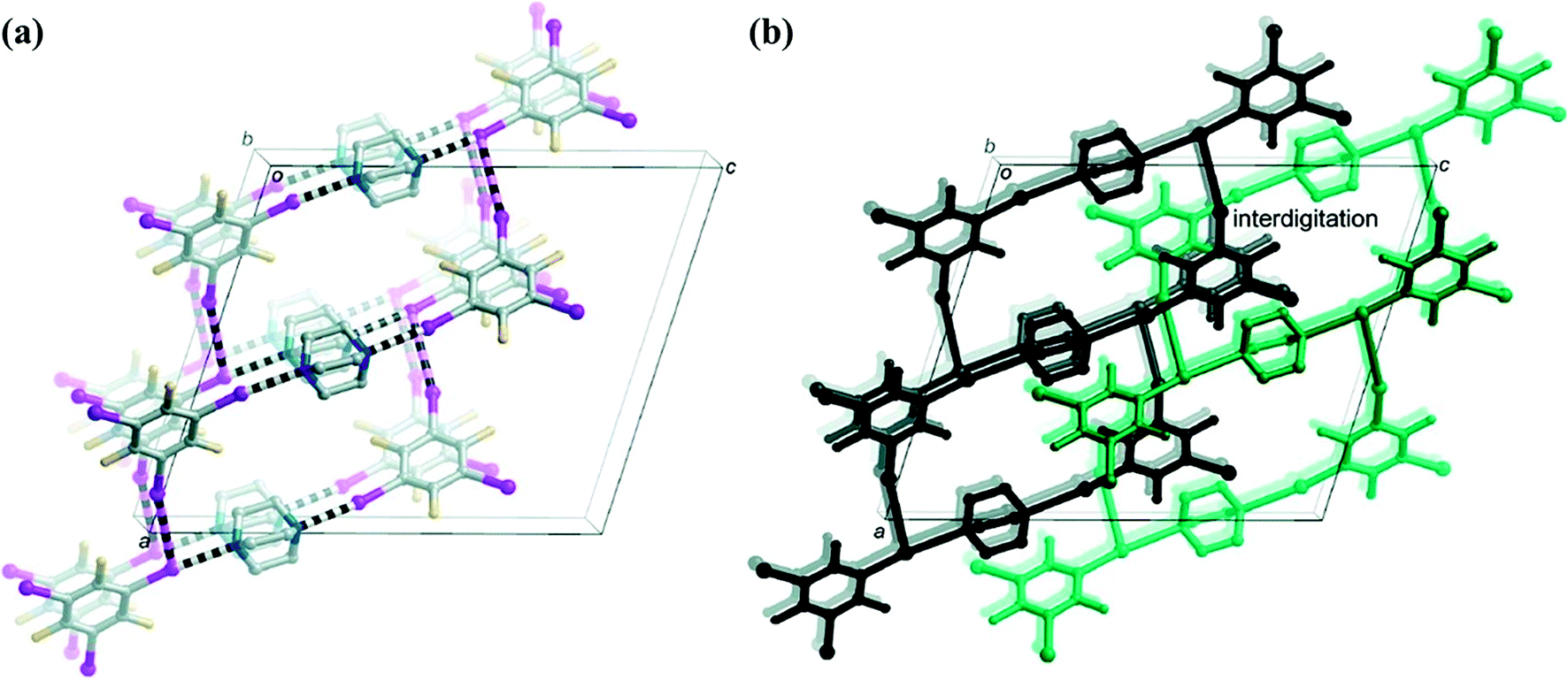 | ||
| Fig. 9 (a) 2D tape-like networks in (1,3,5-TFTIB)2DABCO. (b) Adjacent tapes interdigitate (one black, the other green) via π-stacking interactions. Reprinted with permission from ref. 60. Copyright 2012 The American Chemical Society. | ||
1,3,5-TFTIB exhibited lower-than-expected supramolecular connectivity when co-crystallized with a neutral nitrogen-based halogen-bonding acceptor (DABCO). The likely reason may be the favorable packing arrangements, combined with the possibility that charge donation from the halogen-bonding interaction at two I atoms has an adverse effect on the ability of the third iodine atom to accept a pair of electrons and form the third halogen bond. Such interesting results inspired a search for polymorphs in a different solvent and reactant ratio.
Polymorphous (1,3,5-TFTIB)(DABCO) was obtained by exposing equimolar solutions of 1,3,5-TFTIB and DABCO in chloroform to a saturated n-hexane atmosphere at room temperature.48 In contrast with the polymorph above, it bears a different crystal stacking. An infinite zig-zag chain was found in the co-crystal with a 1:1 ratio, where only two of the three iodine atoms are involved in halogen-bonding interactions. The 1D ribbons are aligned to form distinct 2D sheets interconnected by short C–H⋯F contacts. The layers are stacked in the third dimension with hydrocarbons and perfluorocarbons being segregated.
Replacing DABCO with hexamethylenetetramine (HMTA), an isostructural co-crystal was formed, (1,3,5-TFTIB)(HMTA), which is composed of arcade-like chains in virtue of the geometry of tetramine, with only two of the four HTMA nitrogen atoms being halogen-bonding acceptors.48 A multitude of short C–H⋯F contacts connected such arcade-like chains into unique 2D sheets, which show a striking similarity to its isostructural complex (1,3,5-TFTIB)(DABCO). It is notable that the layers of the DABCO-containing compound are completely planar, whereas (1,3,5-TFTIB)(HMTA) displays undulated sheets. 13C and 19F solid-state NMR was carried out by Bryce's group and offered information on the halogen-bonding chemical environments of (1,3,5-TFTIB)(HMTA), along with GIPAW DFT calculations.57
2.2. N-oxides as halogen-bonding acceptors
N-oxides show the ability to form bifurcated non-covalent bonds due to their high polarization from the charge separation of the N–O bond. On account of the bifurcated angle (donor–O–donor), which is usually close to 120°, Aakeröey and co-workers employed two N-oxides (pyridine-1-oxide and 2,3,5,6-tetramethylpyrazine-1,4-dioxide (TMPD)) as potential halogen-bonding acceptors and 1,3,5-TFTIB as the halogen-bonding donor, which would offer the appropriate geometric complementarity in order to facilitate the assembly of the desired hexagons.61 They used a 1:1 stoichiometry during the solvent-assisted grinding preparation of supramolecular complexes, and indeed obtained 1:1 co-crystals. Both co-crystals contained bifurcated halogen bonds to the N-oxide oxygen atoms with the I⋯O⋯I angles of 117° and nearly 120°, respectively. The halogen-bonding complex formed from mono-N-oxide featured an infinite chain instead of the intended discrete hexagons while the crystalline complex based on bis-N-oxide afforded the desired chain of hexagons (Fig. 10).
 | ||
| Fig. 10 (a) Bifurcated halogen bonds in the co-crystal between 1,3,5-TFTIB and pyridine-1-oxide. (b) Complementarity of angle and geometry leading to fused hexagons in the co-crystal between 1,3,5-TFTIB and TMPD. Reprinted with permission from ref. 61. Copyright 2014 The Royal Society of Chemistry. | ||
With the aim of achieving a higher number of halogen-acceptor units, Raffo et al. attempted to use a 1:9 (1,3,5-TFTIB:pyridine-1-oxide) molar ratio, generating a 1:2 analogue.62 However, it did not exhibit a greater number of C–I⋯O interactions than its analogue discussed above, even if the double N-oxide component is shared. Apart from very minor differences, they are very similar to each other.
2.3. Triphenylphosphine selenide (Ph3PSe) as halogen-bonding acceptor
Co-crystallization of triphenylphosphine selenide (Ph3PSe) and 1,3,5-TFTIB yielded a novel compound (Ph3PSe)(1,3,5-TFTIB), featuring P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Se⋯I–C halogen bonds.63 The co-crystal has two crystallographically distinct PSe environments (Fig. 11). The first selenium atom is surrounded by three iodine atoms from different 1,3,5-TFTIB molecules, forming a one-dimensional zig-zag chain along the b axis. The second one interacts with a single iodine atom, leading to discrete entities in the crystal lattice.
Se⋯I–C halogen bonds.63 The co-crystal has two crystallographically distinct PSe environments (Fig. 11). The first selenium atom is surrounded by three iodine atoms from different 1,3,5-TFTIB molecules, forming a one-dimensional zig-zag chain along the b axis. The second one interacts with a single iodine atom, leading to discrete entities in the crystal lattice.
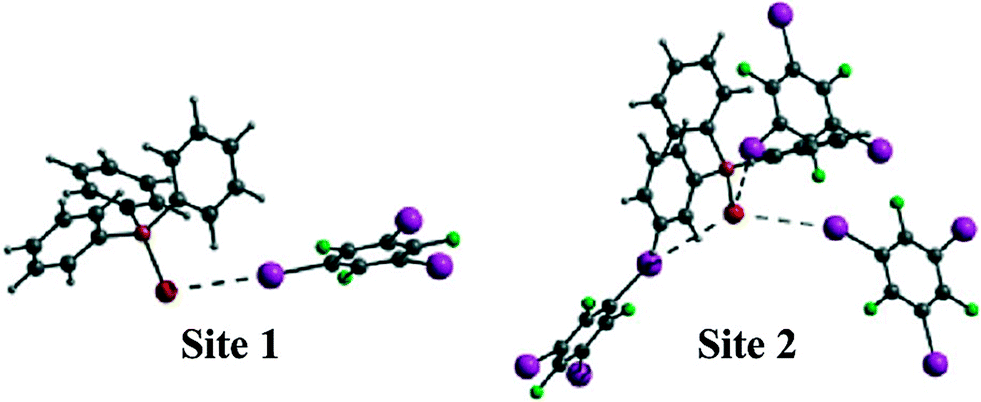 | ||
| Fig. 11 Two crystallographically distinct selenium sites in (Ph3PSe)(1,3,5-TFTIB). Reprinted with permission from ref. 63. Copyright 2014 The Royal Society of Chemistry. | ||
77Se isotopic chemical shifts increase due to halogen bonding with iodine and correlate with the PSe distance, which in turn correlates with the strength of the halogen bond. J(77Se,31P) coupling constants increase in magnitude as the selenium–iodine halogen bonds weaken, and computed J(77Se,31P) improve relative to the experimental values when the iodinated halogen-bonding acceptor is included in the structural model used for calculations, suggesting that J(77Se,31P) are at least in part a characteristic of the halogen bond. The natural localized molecular orbital analysis indicates that contributions from the selenium lone pair orbital tend to dominate both the magnitude and trend in J(77Se,31P), with the selenium–phosphorus bonding orbital being the second largest contributor.
2.4. Anions as the halogen-bonding acceptors
Anion coordination and anion-templated assembly under the control of halogen bonds are still in their infancy but expected to have a bright prospect due to the fundamental role of anions.64–68 Anions are better halogen-bonding acceptors than neutral species, as the increased electron density on the electron-donor site enhances its Lewis basicity, promoting the involvement in a strong and directional halogen bond.69In general, halogen-bonding supramolecular architectures formed by anions are divided into two groups, namely heteromeric two-component and three-component systems.70 In heteromeric two-component systems, the anion serves as the halogen-bonding acceptor and the halogenated organic cation functions as the halogen-bonding donor. The necessity to balance the positive and negative charges greatly affects the number of halogen bonds formed by the anion. In the latter case, the cation actually plays no active role and the halogen-bonding donor is the third neutral component. The three-component systems are better tailored to study the halogen-bonding potential in anion coordination chemistry since there is no other limiting factor in the number, geometry and topology of halogen bonds but the ability of the anion to template the halogen-bonding donor.
In the three-component systems, anions, particularly halides, are readily halogen bonded to the iodinated conjugated molecule such as 1,3,5-TFTIB, affording attractive supramolecular architectures with various connectivities and dimensionalities. Halide ions show preference for the formation of two or three halogen bonds.
2.4.1.1. Metal complex cations as the counterions. The reaction of [Ni(phen)3]X2 (phen = 1,10-phenanthroline; X = Cl− or I−) and 1,3,5-TFTIB resulted in the formation of two co-crystals, [Ni(phen)3][(1,3,5-TFTIB)2Cl2] and [Ni(phen)3][(1,3,5-TFTIB)2I2]·0.5MeOH·1.5H2O.71 The [Ni(phen)3]2+ cations are conserved and arranged into ordered arrays, without usually participating in halogen bonding. In [Ni(phen)3][(1,3,5-TFTIB)2Cl2], the metal complex cations interact through Offset Face-to-Face (OFF) π interactions to form the less common 1D inner OFF zig-zag chain, in which the cleft between the overlapping phen ligands is parallel to the chain's axis of propagation (pink square in Fig. 12b). In the latter case, [Ni(phen)3]2+ cations form a 2D (6,3) net, with a distance of 12.01 Å perpendicularly between each net (Fig. 12d).
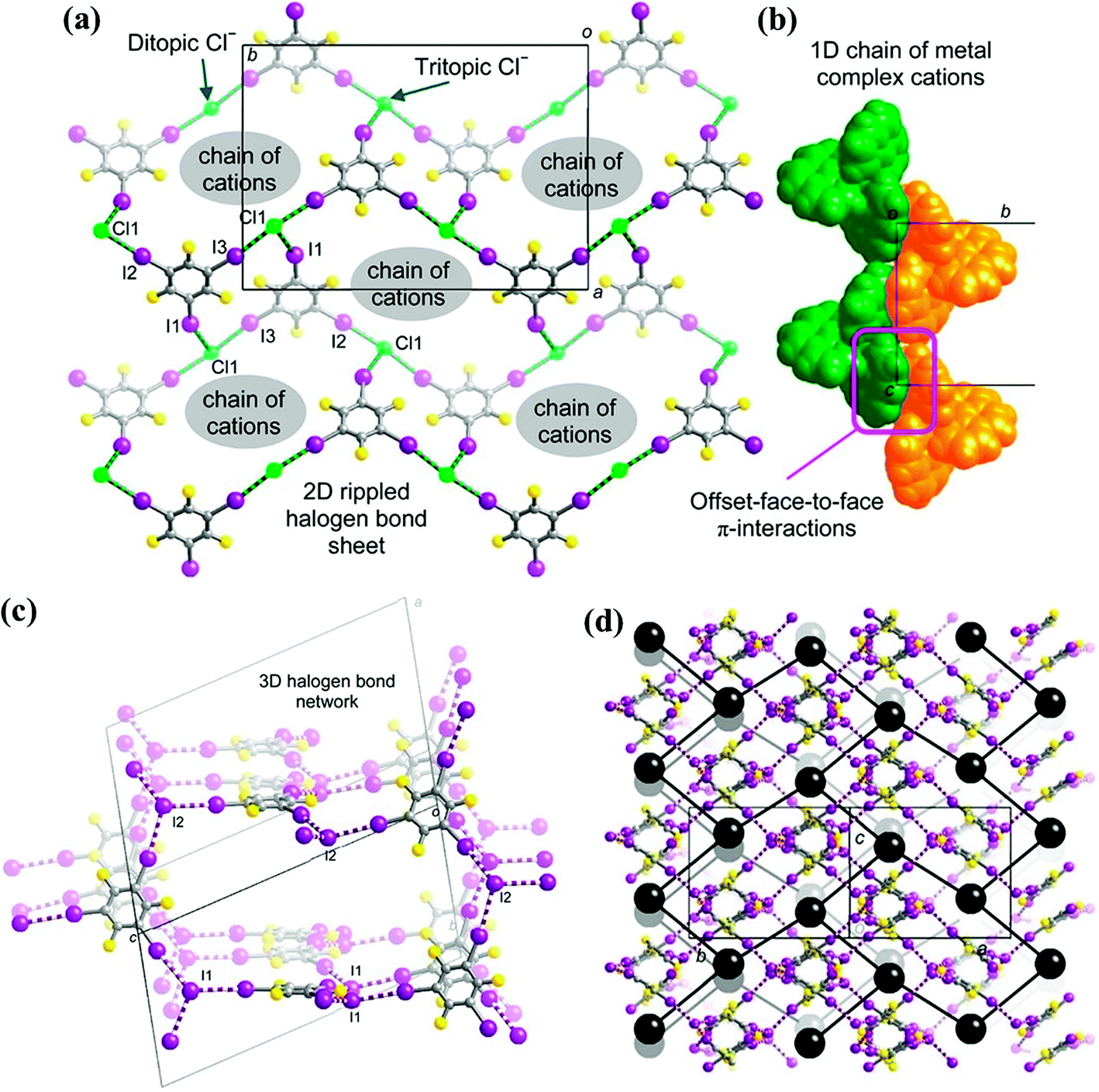 | ||
| Fig. 12 (a) Supramolecular 2D rippled sheet between 1,3,5-TFTIB molecules and Cl− ions in [Ni(phen)3][(1,3,5-TFTIB)2Cl2]. (b) 1D chain of [Ni(phen)3]2+ ions propagating through π–π interactions. (c) A fragment of the 3D halogen-bonding networks between 1,3,5-TFTIB molecules and I− ions in [Ni(phen)3][(1,3,5-TFTIB)2I2]·0.5MeOH·1.5H2O. (d) A perspective view of the lattice highlighting the 2D (6,3) net of [Ni(phen)3]2+ cations (cations abbreviated as black spheres; 2D (6,3) net depicted as black rods). Reprinted with permission from ref. 71. Copyright 2016 The American Chemical Society. | ||
In the two heteromeric three-component systems, halide ions (Cl−, I−) balance the charge of metal complex cations and simultaneously act as halogen-bonding acceptors for the iodine atoms of the 1,3,5-TFTIB donors. Tritopic halide ions and 1,3,5-TFTIB form trigonal nodes, which afford high-dimensional halogen-bonding motifs. In [Ni(phen)3][(1,3,5-TFTIB)2Cl2], unusual rippled 4.82 2D supramolecular sheets are formed between 1,3,5-TFTIB molecules and chloride ions, and cationic chains propagate through channels in the stacked halogen-bonding sheets (Fig. 12a). In the iodide counterpart, two interpenetrating 3D halogen-bonding networks are present, which take the form of a (10,3)-b or three-connected net (Fig. 12c). The 2D (6,3) nets of cations mentioned above are penetrated and linked by the two interpenetrating halogen-bonding networks. Therefore, the nature of halide anions significantly affects the halogen-bonding networks and metal complex motifs. Stronger halogen bonds were expected with the smaller Cl− anion due to its greater charge density while the I− anion forms longer halogen bonds in virtue of its greater size, suggesting that the corresponding halogen-bonding networks may be more flexible in accommodating metal complex cations.
The cryptate K.2.2.2.⊂KI can also serve as a source of naked iodide anions, where K.2.2.2. is 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8,8,8]hexacosane (HDABCOH).72 Single crystal X-ray analysis revealed that both 1,3,5-TFTIB and I− are engaged in three short C–I⋯I− contacts, offering a supramolecular anionic network. The cationic layer is separated from supramolecular anionic layers, with an interlayer distance of 12.209 Å.
2.4.1.2. Onium ions (NR4+, PR4+, SR3+) as the counterions. The Menshutkin reaction has been applied to the synthesis of halogen-bonding supramolecular salts by McMurtrie and co-workers, which occurs between amines and alkyl halides, producing halide anions and quaternary ammonium cations. The in situ generation of 1,3,5-TFTIB and DABCO (in a 2
:3 molar ratio) in a 50:50 mixture of CH2Cl2 and CHCl3 facilitated the formation of a new halogen-bonding network, (DABCO-CH2Cl)3[(1,3,5-TFTIB)2Cl3]·CHCl3, differing significantly from neutral co-crystals stated above.60 In fact, they have attempted using neat CH2Cl2 solvent but failed. Moreover, kinetic studies carried out by Gainza et al. indicate that CHCl3 is essentially unreactive toward tertiary amines at ambient temperature and pressure,73 though the mechanism for the Menshutkin reaction is nucleophilic substitution. However, CHCl3 as the ideal solvent can be utilized to dilute CH2Cl2 for the purpose of Menshutkin reaction.
The chloride ions act as ditopic linear halogen-bonding bridges between 1,3,5-TFTIB molecules where each iodine is involved in one halogen bond, forming a 2D pseudo-trigonal (6,3) framework by charge-assisted C−I⋯Cl− halogen-bonding interactions (Fig. 13a). Three such 2D networks interpenetrate in a rare example of Borromean entanglement by serendipity and offer a supramolecular “tri-net” layer, as depicted in Fig. 13b. The cavities in triply interpenetrated nets are occupied by the cations while CHCl3 molecules reside in the space between the “tri-net” layers.
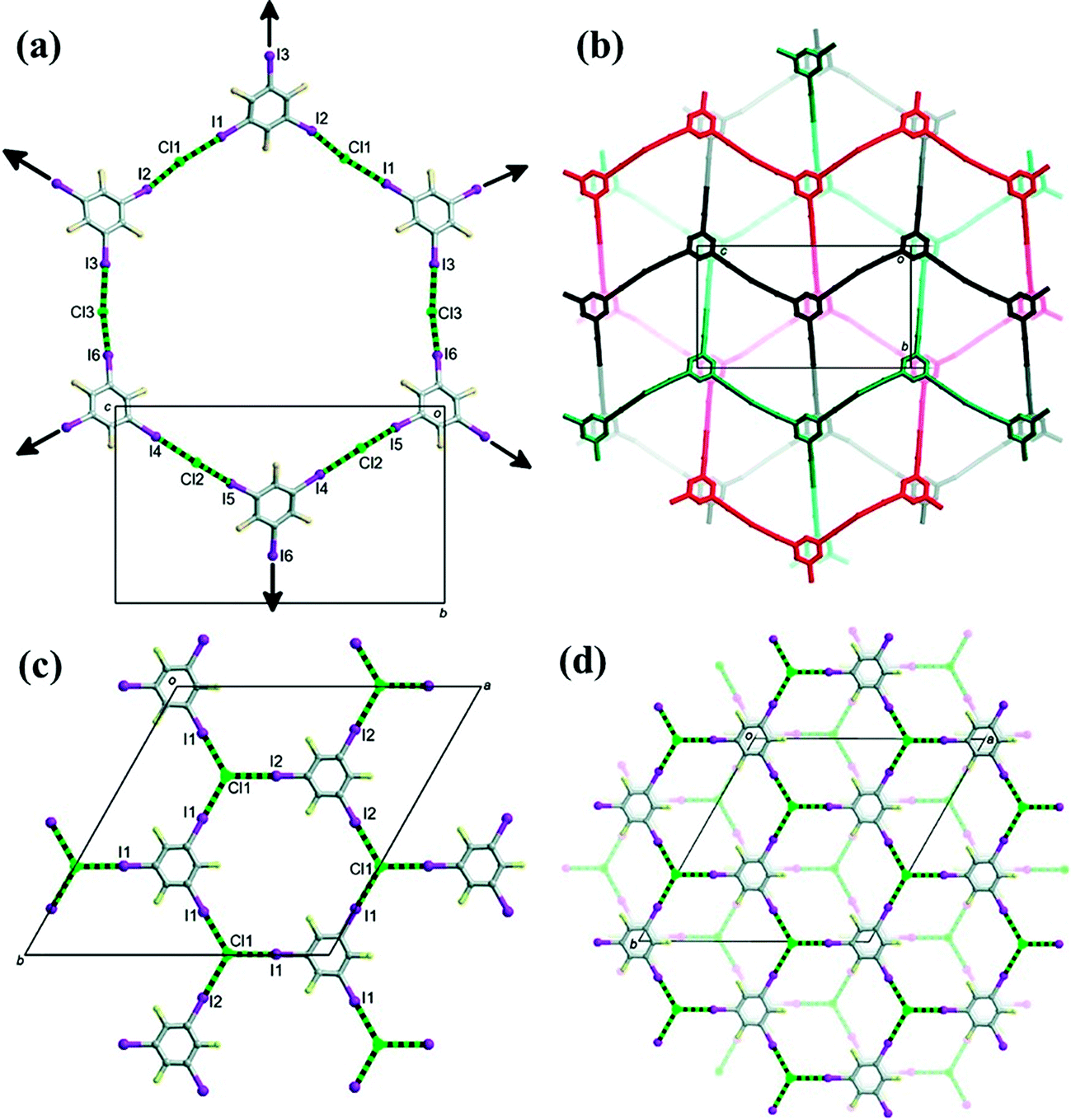 | ||
| Fig. 13 (a) 1,3,5-TFTIB molecules are linked by chloride ions to create a pseudotrigonal 2D network in (1,3,5-TFTIB)2[(DABCO-CH2Cl)Cl]3·CHCl3. (b) Three 2D pseudotrigonal networks interpenetrate in Borromean fashion to create a supramolecular “tri-net” layer. (c) 1,3,5-TFTIB molecules are linked by chloride ions to create 2D nets propagating in the ab plane in (1,3,5-TFTIB)[(TEA-CH2Cl)Cl]. (d) Contiguous nets stack through π–π interactions between 1,3,5-TFTIB molecules. Reprinted with permission from ref. 60. Copyright 2012 The American Chemical Society. | ||
The method was found to be versatile and substitution of DABCO with a different tertiary amine, triethylamine (TEA), gave (TEA-CH2Cl)[(1,3,5-TFTIB)Cl].60 Iodine atoms from a 1,3,5-TFTIB molecule are all halogen bonded to tritopic chloride ions, unlike that in the supramolecular complex discussed above. A 2D (6,3) net with a trigonal crystallographic symmetry has been observed with Cl− and 1,3,5-TFTIB alternating as connecting nodes (Fig. 13c and d). The cations are completely enclosed in the cavities between 2D trigonal halogen-bonding nets.
By comparison, we find that the two products by the Menshutkin reaction both include 1,3,5-TFTIB and Cl− as the supramolecular connectors, and exhibit 2D (6,3) networks. However, the former supramolecular salt contains ditopic Cl− ions and forms interpenetrating nets with Borromean entanglement while Cl− anions are tritopic in the latter case and no interpenetration arises. Hence, there is no doubt that the counter cations with a different size, shape and flexibility make a significant contribution through electrostatic charge balance and space filling although they do not play a direct role in the halogen-bonding interaction. Small TEA-CH2Cl+ cations occupy the well-defined cavities in the tightly connected network. But the larger and less flexible DABCO-CH2Cl+ needs a different halogen-bonding topological framework in which the requirements for space filling are alleviated by interpenetration.
Attempts to extend this work to other onium ions, such as Bu4N+ and Ph4P+, afforded two chlorides ((1,3,5-TFTIB)(n-Bu4NCl) and (1,3,5-TFTIB)(Ph4PCl)·(H2O)0.5), and two bromides ((1,3,5-TFTIB)(n-Bu4NBr) and (1,3,5-TFTIB)(Ph4PBr)).74 The four supramolecular salts are almost isostructural, where each halide anion (Cl−, Br−) is halogen bonded to three I atoms from different 1,3,5-TFTIB molecules, producing a 2D anionic network (Fig. 14), characterized by short I⋯X distances and linear C–I⋯X but acute I⋯X⋯I angles (X = Cl−, Br−). Topological analysis of the electron localization function (ELF) has shown a perfect match between the iodine valence shell electrons, with a “belt”-shape arrangement around the C–I axis, and the spherical halide anions. Moreover, the acute I⋯X⋯I angles are not attributed to any distortion of the halide electron density or any intermolecular bonding I⋯I interaction but rather to favored intermolecular dispersion forces of van der Waals type.
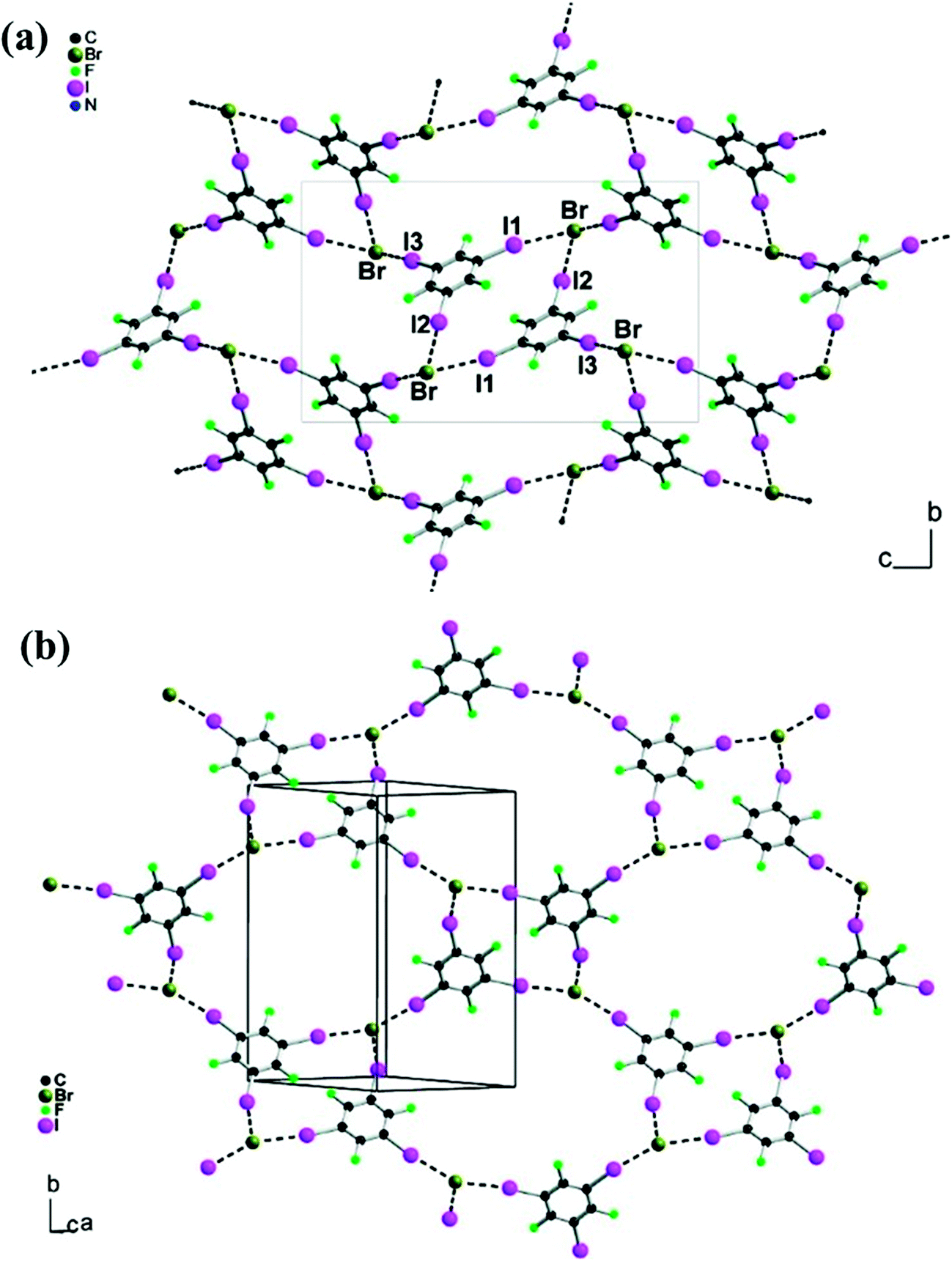 | ||
| Fig. 14 A detailed view of the polymeric anionic [(1,3,5-TFTIB)Br]− network in (1,3,5-TFTIB)(n-Bu4NBr) (a) and (1,3,5-TFTIB)(Ph4PBr) (b). Reprinted with permission from ref. 74. Copyright 2008 The American Chemical Society. | ||
Another bromide, (1,3,5-TFTIB)2(n-Pr4NBr), was reported by Metrangolo's group, which was prepared by slow evaporation of a methanol solution containing n-propyl ammonium bromide as a source of Br− ions and 1,3,5-TFTIB as the halogen-bonding donor.69 The tetradentate Br− anion functions as the halogen-bonding acceptor and bridges four different 1,3,5-TFTIB molecules through C–I⋯Br− interactions with the distances of 3.291 and 3.562 Å. The I⋯Br−⋯I angles are in the range of 70.74–124.47°. In turn, two iodine atoms from 1,3,5-TFTIB generate two halogen bonds with two different bromide ions while another iodine atom not involved in C–I⋯Br− interactions is engaged in short contacts with fluorine atoms. As a consequence of these robust halogen-bonding interactions, a 2D (4,4) supramolecular network forms, in which Br− anions sit at the nodes of a highly distorted square defined by four Br− anions (vertexes) and four 1,3,5-TFTIB molecules (sides) (Fig. 15).
 | ||
| Fig. 15 The 2D (4,4) supramolecular network between 1,3,5-TFTIB molecules and Br− anions in (1,3,5-TFTIB)2(n-Pr4NBr). Reprinted with permission from ref. 69. Copyright 2010 Elsevier. | ||
When it comes to the iodine ion, a series of supramolecular salts were prepared, namely (1,3,5-TFTIB)(Me3SI), (1,3,5-TFTIB)(Et4NI), (1,3,5-TFTIB)2(n-Pr4NI)·CH2Cl2, (1,3,5-TFTIB)(n-Bu4NI)·(CH2Cl2)0.5, (1,3,5-TFTIB)(Et4PI) and (1,3,5-TFTIB)(Ph4PI)·CHCl3, with the counterions ranging from sulfonium to ammonium and phosphonium.72 In all cases, a 1,3,5-TFTIB molecule and a naked I− anion work as a tridentate halogen-bonding donor and acceptor, respectively.
To be specific, in (1,3,5-TFTIB)(Me3SI), the trigonal coordination profile of a 1,3,5-TFTIB molecule and an iodine ion results in the formation of a honeycomb-like (6,3) supramolecular anionic network via C–I⋯I− interactions with distances that vary from 3.385 to 3.508 Å. Planar honeycomb-like 2D layers are 3.608 Å apart from each other. The small Me3S+ cations sit perfectly in the centre of the hexagonal frameworks (Fig. 16a), which are pinned by weak H⋯F contacts and electrostatic interactions, slightly deforming the hexagonal frameworks with I⋯I−⋯I angles in the range of 110.25–139.41°.
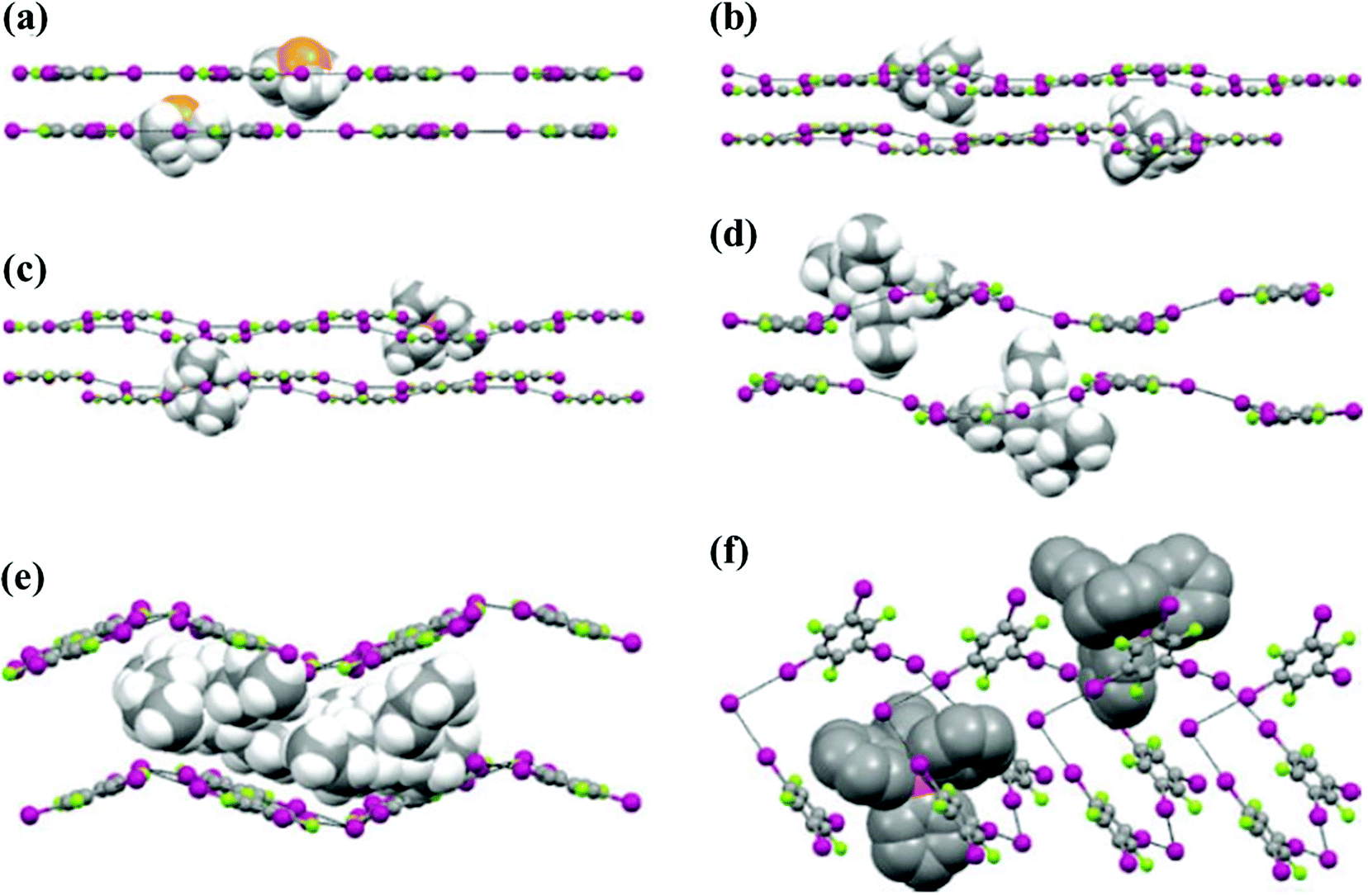 | ||
| Fig. 16 Overall packing of heteromeric crystals. (a) (1,3,5-TFTIB)(Me3SI), (b) (1,3,5-TFTIB)(Et4NI), (c) (1,3,5-TFTIB)2(n-Pr4NI)·CH2Cl2, (d) (1,3,5-TFTIB)(n-Bu4NI)·(CH2Cl2)0.5, (e) (1,3,5-TFTIB)(Et4PI) and (f) (1,3,5-TFTIB)(Ph4PI)·CHCl3. Anionic networks are in ball-and-stick style while cations are in spacefill style. Reprinted with permission from ref. 72. Copyright 2008 The Royal Society of Chemistry. | ||
With the purpose of identifying the critical cation size in relation to the ability of the 1,3,5-TFTIB–I− system to form (6,3) networks, Metrangolo's group challenged 1,3,5-TFTIB with various ammonium iodides possessing larger and larger cations (Et4NI, n-Pr4NI and n-Bu4NI). In all of the three cases, tridentate 1,3,5-TFTIB and I− produce 2D supramolecular anionic networks through short C–I⋯I− contacts. Interestingly, heteromeric crystals obtained from Et4NI and n-Pr4NI both display the (6,3) network topology, while the one achieved from n-Bu4NI presents a different network. Although Et4N+ cations still sit perfectly in the centre of the hexagonal frameworks (Fig. 16b), n-Pr4N+ cations are moved slightly out of the hexagonal frameworks (Fig. 16c) and n-Bu4N+ cations are moved even further out of the 2D layer so that alternating anionic and cationic layers compose its overall crystal packing (Fig. 16d). As the cation size increases, the anionic networks become more corrugated and their distance enlarges, moving from 4.529 to 5.549 and 7.986 Å. Accordingly, the upper limit of the cation size for preventing the 1,3,5-TFTIB–I− system from forming (6,3) networks seems to lie between the dimensions of n-Pr4N+ and n-Bu4N+ cations.
As far as the heteromeric (1,3,5-TFTIB)(Et4PI) is concerned, the halogen-bonding pattern and overall topology are strikingly similar to that of its ammonium analogue, exhibiting a supramolecular anionic network with the (6,3) topology. The centres of the hexagonal frameworks are occupied by the Et4P+ cations (Fig. 16e and 17). Since the Et4P+ ion is larger than Et4N+ cation, the separation between layers of anionic networks is slightly greater (4.601 Å).
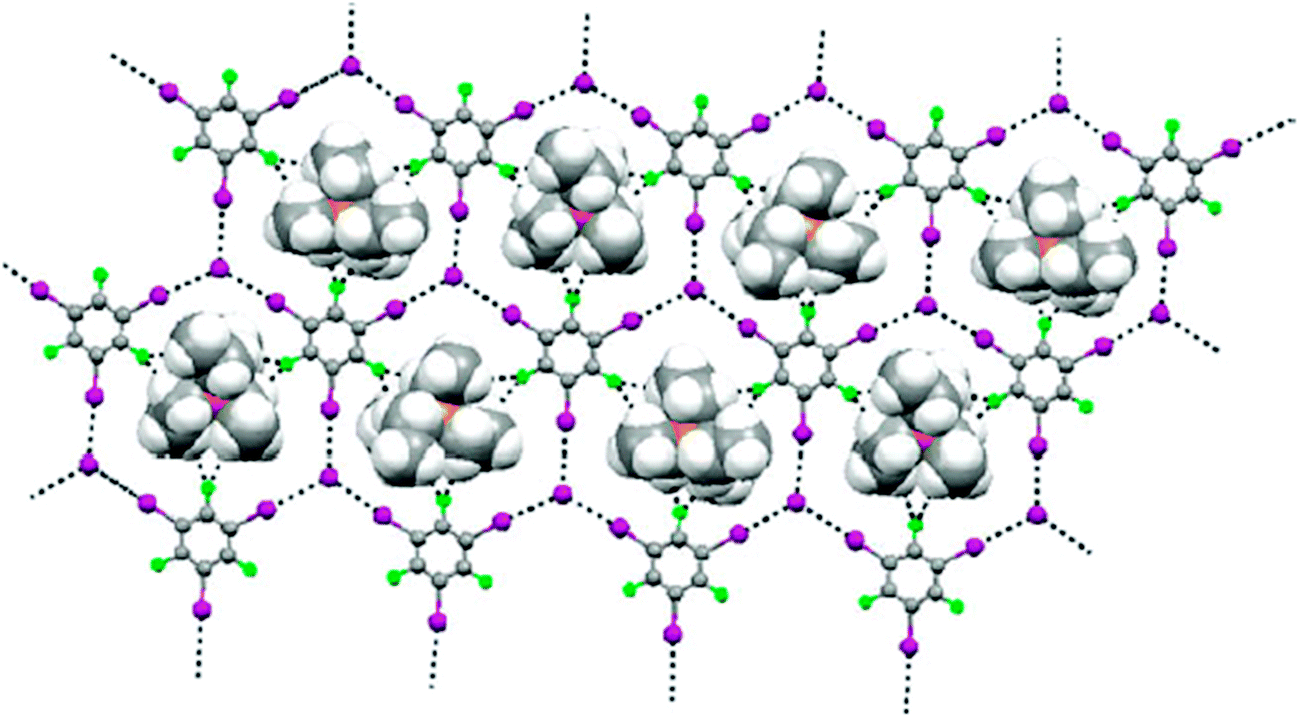 | ||
| Fig. 17 The Et4P+ cations are accommodated in the hexagonal frameworks with the (6,3) topology between 1,3,5-TFTIB molecules and I− anions in (1,3,5-TFTIB)(Et4PI). Reprinted with permission from ref. 72. Copyright 2008 The Royal Society of Chemistry. | ||
Replacing Et4P+ with a large Ph4P+ cation, (1,3,5-TFTIB)(Ph4PI)·CHCl3 was formed, where the (6,3) topology has been blocked and opens up into a hexagonal helical network, mimicking the honeycomb-like structure and accommodating the bulky tetraphenylphosphonium cations (Fig. 16f). Polymorphic (1,3,5-TFTIB)4(Ph4PI)3·MeOH crystallized from a methanol solution, refined as a perfect inversion twin.75 Differing from the cases mentioned above, 1,3,5-TFTIB serves as the bi- or tridentate halogen-bonding donor and the iodide anion functions as the bi-, tetra- or pentadentate halogen-bonding acceptor.
In brief, (6,3) anionic network is robust enough to resist disruption due to small changes in the cation for Me3S+, Et4N+, Pr4N+ and Et4P+, but is destroyed when larger cations are incorporated such as Bu4N+ and Ph4P+. Consequently, honeycomb-like anionic networks are obtained if the cation nicely fits in the hexagonal frameworks; if not, supramolecular architectures with different topologies form to accommodate the counterions, with the tridentate character of halogen-bonding donors and acceptors being maintained.
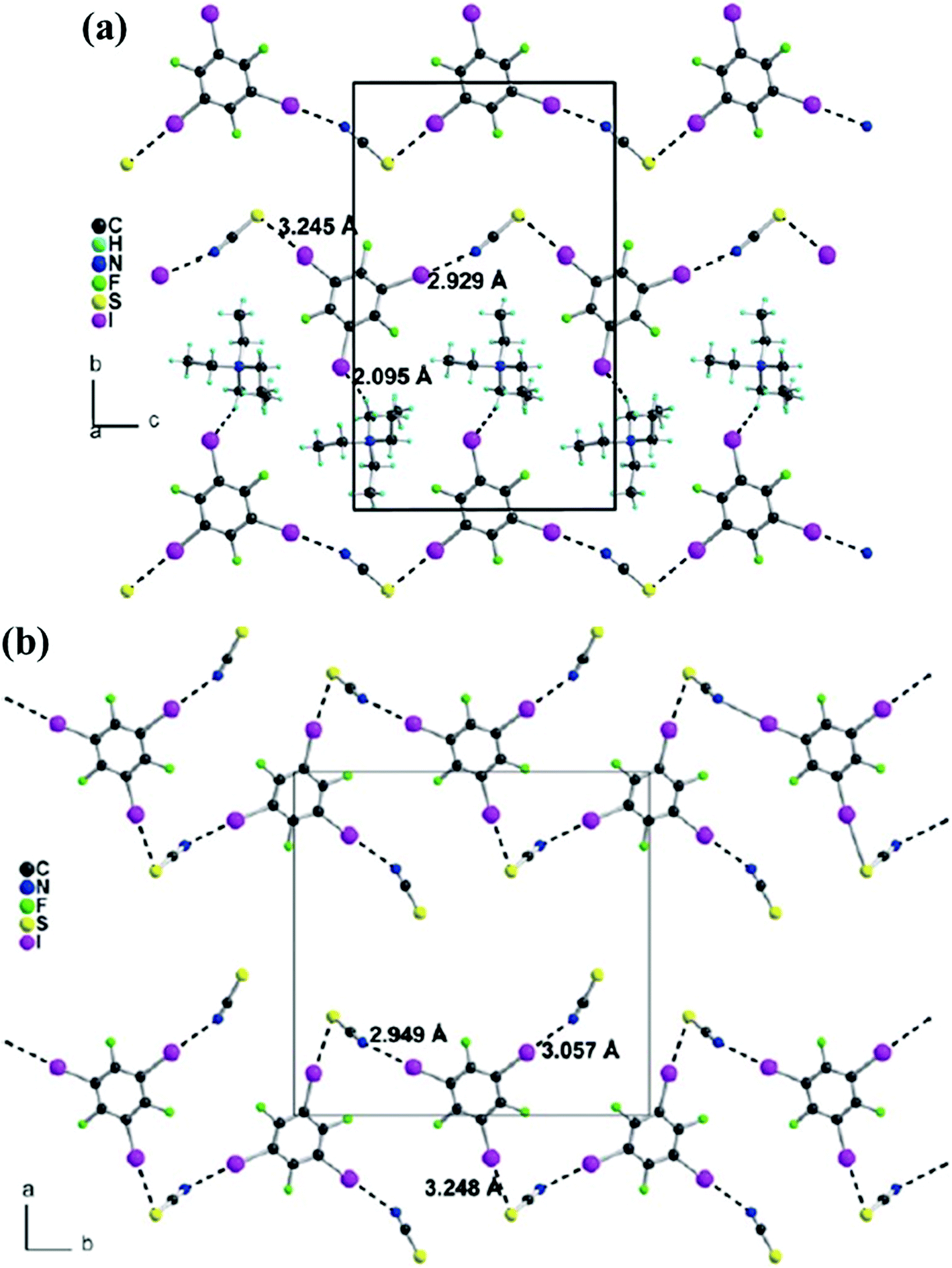 | ||
| Fig. 18 (a) Halogen-bonding chains in (Et4NSCN)(1,3,5-TFTIB). (b) Halogen-bonding chains between 1,3,5-TFTIB molecules and SCN− anions in (n-Bu4NSCN)2(1,3,5-TFTIB). The n-Bu4N+ cation has been omitted for clarity. Reprinted with permission from ref. 76. Copyright 2010 The Royal Society of Chemistry. | ||
The corresponding tetrabutylammonium salt, (n-Bu4NSCN)2(1,3,5-TFTIB), is found to crystallize with a different stoichiometry. The three iodine atoms of the 1,3,5-TFTIB molecule are all involved in halogen bonds, in which two of them with one SCN− anion to form interesting chains through C–I⋯S interactions running along the b axis while the third one is linked to the nitrogen atom of another SCN− anion with a slightly longer C–I⋯N distance (Fig. 18b). Especially deserving to be mentioned, the SCN− anion features two kinds of bonding modes, namely terminal and bridging. All in all, the bitopic character of the SCN− anion, with both the S and N atoms engaged in halogen-bonding interactions, has been revealed by the analyses of the two crystal structures combined with theoretical calculations.
3. Conclusion
In this review, we have attempted to outline the efforts to obtain new multicomponent supramolecular complexes by the co-crystallization between 1,3,5-TFTIB and various halogen-bonding acceptors, ranging from neutral Lewis bases (nitrogen-containing heterocycles, N-oxides, and triphenylphosphine selenide (Ph3PSe)) to anions (halide ions and thiocyanate ion). Among them, nitrogen-containing heterocycles have been widely studied, including six-membered rings (pyridine, pyrimidine and pyrazine) and five-membered rings (imidazole, oxadiazole, thiadiazole and triazole). The examples presented here illustrate that halogen bonds play a major role in the intermolecular recognition and self-assembly processes, showing a great variety of impressive supramolecular architectures (e.g. V-shaped trimer, 1D zig-zag chain, 2D tape-like network, rippled 4.82 2D sheet, 2D (6,3) network, 2D (4,4) network and interpenetrating 3D network). Besides the crystallographic method, electrostatic potential and chemical shifts have been utilized to explain halogen-bonding interactions.1,3,5-TFTIB is a potentially tritopic halogen-bonding donor with three-fold symmetry, but it forms the more-usual two halogen bonds with neutral acceptors despite the existence of the case of one (for example, 1-(pyridin-3-ylmethyl)-benzimidazole) or three halogen bonds (such as 4-dimethylaminopyridine). This reluctance to reach a full “coordination” was attributed to a deactivation of the iodine atoms upon successive interactions with one, two or three Lewis bases. Even so, if the crystal packing is favorable, 1,3,5-TFTIB may act as a tritopic halogen-bonding linker with neutral Lewis bases. As for anionic species, all the three iodine atoms can simultaneously participate in halogen-bonding interactions, resulting in 2D trigonal networks. An increased electron density in the anion enhances its Lewis basicity thus promoting the involvement of three iodine atoms from 1,3,5-TFTIB in the simultaneous formation of three halogen bonds.
Although the use of 1,3,5-TFTIB in the co-crystallization research field has been nearly for ten years, we believe new supramolecular architectures will appear in the near future. Exploration of functional materials, such as efficient optoelectronic materials, has only just begun, and it is worth looking forward. Even less is known about surface molecular engineering. This review is anticipated to provide useful information and stimulate further research into this fascinating class of halogen-bonding supramolecular complexes. Furthermore, a review on the linear ditopic 1,2,4,5-tetrafluoro-3,6-diiodobenzene (TFDIB) is now in progress and will be reported in due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Basic Research Program of China (973 Program, Grant No. 2015CB932200), the Synergetic Innovation Center for Organic Electronics and Information Displays, and the Jiangsu Planned Projects for Postdoctoral Research Funds (1601042B).References
- D. B. Amabilino, D. K. Smith and J. W. Steed, Chem. Soc. Rev., 2017, 46, 2404–2420 RSC
.
- I. V. Kolesnichenko and E. V. Anslyn, Chem. Soc. Rev., 2017, 46, 2385–2390 RSC
- L. K. S. von Krbek, C. A. Schalley and P. Thordarson, Chem. Soc. Rev., 2017, 46, 2622–2637 RSC
- K. S. Mali, N. Pearce, S. De Feyter and N. R. Champness, Chem. Soc. Rev., 2017, 46, 2520–2542 RSC
- J. Zhou, J. Li, X. Du and B. Xu, Biomaterials, 2017, 129, 1–27 CrossRef CAS PubMed
- J. W. Steed, Trends Pharmacol. Sci., 2013, 34, 185–193 CrossRef CAS PubMed
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed
- D. P. Yan and D. G. Evans, Mater. Horiz., 2014, 1, 46–57 RSC
- Y. Wang, W. G. Zhu, H. L. Dong, X. T. Zhang, R. J. Li and W. P. Hu, Top. Curr. Chem., 2016, 374 Search PubMed
- W. G. Zhu, L. Y. Zhu, Y. Zou, Y. S. Wu, Y. G. Zhen, H. L. Dong, H. B. Fu, Z. X. Wei, Q. Shi and W. P. Hu, Adv. Mater., 2016, 28, 5954–5962 CrossRef CAS PubMed
- W. G. Zhu, L. Y. Zhu, L. J. Sun, Y. G. Zhen, H. L. Dong, Z. X. Wei and W. P. Hu, Angew. Chem., Int. Ed., 2016, 55, 14023–14027 CrossRef CAS PubMed
- D. J. Berry and J. W. Steed, Adv. Drug Delivery Rev., 2017 DOI:10.1016/j.addr.2017.03.003
- D. P. Yan, Chem. – Eur. J., 2015, 21, 4880–4896 CrossRef CAS PubMed
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775–2825 CrossRef CAS PubMed
- S. E. Wheeler, T. J. Seguin, Y. Guan and A. C. Doney, Acc. Chem. Res., 2016, 49, 1061–1069 CrossRef CAS PubMed
- C. Sutton, C. Risko and J.-L. Bredas, Chem. Mater., 2016, 28, 3–16 CrossRef CAS
-
P. Metrangolo, G. Resnati, T. Pilati and S. Biella, Halogen Bonding: Fundamentals and Applications (Struct. Bond.), Springer, Berlin, Heidelberg, 2008, vol. 126, pp. 105–136 Search PubMed
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed
-
C. B. Aakeröy and C. L. Spartz, Halogen Bonding I: Impact on Materials Chemistry and Life Sciences (Top. Curr. Chem.), Springer, Switzerland, 2015, vol. 358, pp. 155–182 Search PubMed
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed
- L. P. Wolters, P. Schyman, M. J. Pavan, W. L. Jorgensen, F. M. Bickelhaupt and S. Kozuch, Comput. Mol. Biosci., 2014, 4, 523–540 CrossRef CAS
-
M. H. Kolar, P. Deepa, H. Ajani, A. Pecina and P. Hobza, Halogen Bonding II: Impact on Materials Chemistry and Life Sciences (Top. Curr. Chem.), Springer, Switzerland, 2015, vol. 359, pp. 1–26 Search PubMed
- H. Wang, W. Wang and W. J. Jin, Chem. Rev., 2016, 116, 5072–5104 CrossRef CAS PubMed
-
A. C. Legon, Halogen Bonding: Fundamentals and Applications (Struct. Bond.), Springer, Berlin, Heidelberg, 2008, vol. 126, pp. 17–64 Search PubMed
- A. C. Legon, Phys. Chem. Chem. Phys., 2010, 12, 7736–7747 RSC
- J. Lieffrig, O. Jeannin and M. Fourmigué, J. Am. Chem. Soc., 2013, 135, 6200–6210 CrossRef CAS PubMed
- B. Li, S.-Q. Zang, L.-Y. Wang and T. C. W. Mak, Coord. Chem. Rev., 2016, 308, 1–21 CrossRef CAS
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS PubMed
-
X. Pang and W. J. Jin, Halogen Bonding II: Impact on Materials Chemistry and Life Sciences (Top. Curr. Chem.), Springer, Switzerland, 2015, vol. 359, pp. 115–146 Search PubMed
- G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati, M. Sansotera and G. Terraneo, Chem. Soc. Rev., 2010, 39, 3772–3783 RSC
-
A. V. Jentzsch and S. Matile, Halogen Bonding I: Impact on Materials Chemistry and Life Sciences (Top. Curr. Chem.), Springer, Berlin, Heidelberg, 2015, vol. 358, pp. 205–240 Search PubMed
- M. J. Langton, S. W. Robinson, I. Marques, V. Felix and P. D. Beer, Nat. Chem., 2014, 6, 1039–1043 CrossRef CAS PubMed
- A. Brown and P. D. Beer, Chem. Commun., 2016, 52, 8645–8658 RSC
- G. Cavallo, G. Terraneo, A. Monfredini, M. Saccone, A. Priimagi, T. Pilati, G. Resnati, P. Metrangolo and D. W. Bruce, Angew. Chem., Int. Ed., 2016, 55, 6300–6304 CrossRef CAS PubMed
- P. Metrangolo, Y. Carcenac, M. Lahtinen, T. Pilati, K. Rissanen, A. Vij and G. Resnati, Science, 2009, 323, 1461–1464 CrossRef CAS PubMed
- L. Meazza, J. A. Foster, K. Fucke, P. Metrangolo, G. Resnati and J. W. Steed, Nat. Chem., 2013, 5, 42–47 CrossRef CAS PubMed
-
P. S. Ho, Halogen Bonding I: Impact on Materials Chemistry and Life Sciences (Top. Curr. Chem.), Springer, Berlin, Heidelberg, 2015, vol. 358, pp. 241–276 Search PubMed
- S. Q. Jiang, L. J. Zhang, D. B. Cui, Z. Q. Yao, B. Gao, J. P. Lin and D. Z. Wei, Sci. Rep., 2016, 6 Search PubMed
- P. Metrangolo and G. Resnati, Science, 2008, 321, 918–919 CrossRef CAS PubMed
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS
-
G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Halogen Bonding I: Impact on Materials Chemistry and Life Sciences (Top. Curr. Chem.), Springer, Switzerland, 2015, vol. 358, pp. 1–18 Search PubMed
- N. Ramasubbu, R. Parthasarathy and P. Murray-Rust, J. Am. Chem. Soc., 1986, 108, 4308–4314 CrossRef CAS
- P. Politzer, P. Lane, M. C. Concha, Y. Ma and J. S. Murray, J. Mol. Model., 2007, 13, 305–311 CrossRef CAS PubMed
- P. Metrangolo, G. Resnati, T. Pilati, R. Liantonio and F. Meyer, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1–15 CrossRef CAS
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed
- A. C. B. Lucassen, A. Karton, G. Leitus, L. J. W. Shimon, J. M. L. Martin and M. E. van der Boom, Cryst. Growth Des., 2007, 7, 386–392 CAS
- J.-L. Syssa-Magale, K. Boubekeur, J. Leroy, L.-M. Chamoreau, C. Fave and B. Schöellhorn, CrystEngComm, 2014, 16, 10380–10384 RSC
- W. G. Zhu, R. H. Zheng, Y. G. Zhen, Z. Y. Yu, H. L. Dong, H. B. Fu, Q. Shi and W. P. Hu, J. Am. Chem. Soc., 2015, 137, 11038–11046 CrossRef CAS PubMed
- Y. Liu, S. Ma, B. Xu and W. Tian, Faraday Discuss., 2017, 196, 219–229 RSC
- M. Vartanian, A. C. Lucassen, L. J. Shimon and M. E. van der Boom, Cryst. Growth Des., 2008, 8, 786–790 CAS
- C. B. Aakeröy, T. K. Wijethunga and J. Desper, J. Mol. Struct., 2014, 1072, 20–27 CrossRef
- C. B. Aakeröy, T. K. Wijethunga, J. Desper and M. Đaković, Cryst. Growth Des., 2016, 16, 2662–2670 Search PubMed
- L. C. Roper, C. Prasang, V. N. Kozhevnikov, A. C. Whitwood, P. B. Karadakov and D. W. Bruce, Cryst. Growth Des., 2010, 10, 3710–3720 CAS
- P. I. Hidalgo, S. Leal, C. A. Jimenez, E. Vohringer-Martinez, B. Herrera, J. Pasan, C. Ruiz-Perez and D. W. Bruce, CrystEngComm, 2016, 18, 42–47 RSC
- B. Ji, W. Wang, D. Deng, Y. Zhang, L. Cao, L. Zhou, C. Ruan and T. Li, CrystEngComm, 2013, 15, 769–774 RSC
- P. M. J. Szell, S. A. Gabriel, R. D. D. Gill, S. Y. H. Wan, B. Gabidullin and D. L. Bryce, Acta Crystallogr., Sect. C: Struct. Chem., 2017, 73, 157–167 CAS
- Q.-N. Zheng, X.-H. Liu, T. Chen, H.-J. Yan, T. Cook, D. Wang, P. J. Stang and L.-J. Wan, J. Am. Chem. Soc., 2015, 137, 6128–6131 CrossRef CAS PubMed
- M. D. Perera, J. Desper, A. S. Sinha and C. B. Aakeröy, CrystEngComm, 2016, 18, 8631–8636 RSC
- M. C. Pfrunder, A. S. Micallef, L. Rintoul, D. P. Arnold, K. J. Davy and J. McMurtrie, Cryst. Growth Des., 2012, 12, 714–724 CAS
- C. B. Aakeröy, T. K. Wijethunga and J. Desper, CrystEngComm, 2014, 16, 28–31 RSC
- P. A. Raffo, F. D. Cukiernik and R. F. Baggio, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 84–U257 CAS
- J. Viger-Gravel, J. E. Meyer, I. Korobkov and D. L. Bryce, CrystEngComm, 2014, 16, 7285–7297 RSC
- N. Gimeno and R. Vilar, Coord. Chem. Rev., 2006, 250, 3161–3189 CrossRef CAS
- H. T. Chifotides and K. R. Dunbar, Acc. Chem. Res., 2013, 46, 894–906 CrossRef CAS PubMed
- G. T. Spence and P. D. Beer, Acc. Chem. Res., 2013, 46, 571–586 CrossRef CAS PubMed
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed
- P. A. Gale, J. T. Davis and R. Quesada, Chem. Soc. Rev., 2017, 46, 2497–2519 RSC
- G. Cavallo, S. Biella, J. Lu, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, J. Fluorine Chem., 2010, 131, 1165–1172 CrossRef CAS
- P. Metrangolo, T. Pilati, G. Terraneo, S. Biella and G. Resnati, CrystEngComm, 2009, 11, 1187–1196 RSC
- M. C. Pfrunder, A. S. Micallef, L. Rintoul, D. P. Arnold and J. McMurtrie, Cryst. Growth Des., 2016, 16, 681–695 CAS
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Chem. Commun., 2008, 1635–1637 RSC
- A. H. Gainza, Int. J. Chem. Kinet., 2004, 36, 500–509 CrossRef CAS
- S. Triguero, R. Llusar, V. Polo and M. Fourmigue, Cryst. Growth Des., 2008, 8, 2241–2247 CAS
- G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, o865–866 CAS
- P. Cauliez, V. Polo, T. Roisnel, R. Llusar and M. Fourmigue, CrystEngComm, 2010, 12, 558–566 RSC
Footnote |
| † X.-H. Ding and C.-J. Ou contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |