
Selective adsorption behaviour of carbon dioxide in OH-functionalized metal–organic framework materials†
Jinjie
Qian
 *ab,
Jinni
Shen
b,
Qipeng
Li
b,
Yue
Hu
*a and
Shaoming
Huang
*a
*ab,
Jinni
Shen
b,
Qipeng
Li
b,
Yue
Hu
*a and
Shaoming
Huang
*a
aCollege of Chemistry and Materials Engineering, Wenzhou University, Wenzhou 325035, PR China. E-mail: jinjieqian@wzu.edu.cn; yuehu@wzu.edu.cn; smhuang@wzu.edu.cn; Tel: +86 577 88373064
bState Key Laboratory of Structure Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, China
First published on 17th August 2017
Abstract
The theoretically optimal adsorption locations in hydroxyl (OH)-decorated metal–organic frameworks show that the captured carbon dioxide (CO2) molecules interact with the cis-μ2-OH groups in an end-on mode, which shows a moderate to weak hydrogen bond. The experimental isotherms and ideal adsorption solution theory (IAST) calculations show the high selectivity of CO2 for nitrogen at 273, 283 and 295 K and 1.0 bar for three types of OH-appended isostructures.
The efficient capture and conversion of carbon dioxide (CO2) is of great importance in realizing a carbon-neutral energy cycle and low-carbon society.1 Over a long period of time, humans have become heavily reliant on fossil fuels in agriculture and industry, which is widely believed to present the major source for CO2 emissions, thus leading to the rise of temperatures across the planet, from which a new kind of increasingly serious threat: global warming, may eventually have to be faced.2 Although several developments of techniques for the capture of CO2 have been developed, for example, effective and efficient CO2 adsorption in alkylamine solutions, there are still a series of issues related to cost performance, storage, transportation, and safety with this.3 Therefore, the strong driving force has long been to use alternative high performance strategies to efficiently remove CO2.
Currently, microporous metal–organic frameworks (MOFs), a subclass of crystalline hybrid polymers constructed from metallic cations/clusters and organic ligands, are emerging as one of the most promising candidates for CO2 capture because of their tunable structure, diverse topology, different chemical compositions of the micropores as well as the extra large specific surface area.4 Within the field of small molecule adsorption, a large amount of research has been focused on the mutual interaction mechanism between the MOF framework and the entrapped small molecule guests, especially for non-polar linear CO2 molecule, thus discovering new functional materials with better performance. Recently, the group of Ibarra has demonstrated that alcohol confinement within a previously reported water-stable InOF-1,5 built from octahedral indium(III) hydroxide-biphenyltetracarboxylic acid [In2(OH)2(BPTC)] chain extended by BPTC ligands, can significantly promote CO2 capture capacity with an approximately 3.6-fold increase of performance (from 3.8 to 13.7 mmol g−1) at only 1.0 bar and 30 °C.6 In this case, more suitable narrow pore sizes which can effectively accommodate the small guest molecules and thus provide a strong overlapping potential have also been confirmed to enhance the CO2 capacity as well as the sorption affinity.6 In this respect, it is also necessary to predict and visualize the CO2 positions in the hydroxyl (OH)-decorated pore environment within the MOF materials using theoretical calculations and experimental tests.
It has been learned that high capacity storage in MOF materials can be achieved by the introduction of open metal sites (OMS),7a–c where there exists a strong CO2 binding strength which forms the Mn+⋯C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)2 bond because of the direct interaction between CO2 and the coordinately unsaturated metal centers, causing irreversible physisorption and permanent loss of OMS activity. Fig. 1 shows that traditional alkylamine solutions, alkylamine-appended as well as polar amine-/sulfonate-/ketone-appended MOF materials7d–h can be extensively utilized for the storage and separation of CO2, but few examples are available showing that OH-functionalized MOF materials can effectively remove CO2 because of the limited structural stability of the crystal compounds. In this paper, the optimization of CO2 molecule sites in the InOF-1 framework using density functional theory (DFT) calculations, and a CO2 adsorption behaviour comparison with an aluminium-/gallium-(Al/Ga)-based isostructural framework (AlOF-1 and GaOF-1, respectively), is reported, which confirms that all the OH-appended materials possess high CO2 capacity and selectivity towards nitrogen (N2) at 273 K, 283 K, and 295 K. Furthermore, it was found that AlOF-1 outperforms in CO2 uptake because of the large binding energy used to structurally form AlOF-1·4CO2. More importantly, a high selectivity of CO2/N2 at 273–295 K and 1.0 bar, and a large heat of CO2 adsorption can be achieved at zero coverage (20.37 kJ mol−1for AlOF-1, 18.31 kJ mol−1 for GaOF-1 and 11.98 kJ mol−1 for InOF-1).
O)2 bond because of the direct interaction between CO2 and the coordinately unsaturated metal centers, causing irreversible physisorption and permanent loss of OMS activity. Fig. 1 shows that traditional alkylamine solutions, alkylamine-appended as well as polar amine-/sulfonate-/ketone-appended MOF materials7d–h can be extensively utilized for the storage and separation of CO2, but few examples are available showing that OH-functionalized MOF materials can effectively remove CO2 because of the limited structural stability of the crystal compounds. In this paper, the optimization of CO2 molecule sites in the InOF-1 framework using density functional theory (DFT) calculations, and a CO2 adsorption behaviour comparison with an aluminium-/gallium-(Al/Ga)-based isostructural framework (AlOF-1 and GaOF-1, respectively), is reported, which confirms that all the OH-appended materials possess high CO2 capacity and selectivity towards nitrogen (N2) at 273 K, 283 K, and 295 K. Furthermore, it was found that AlOF-1 outperforms in CO2 uptake because of the large binding energy used to structurally form AlOF-1·4CO2. More importantly, a high selectivity of CO2/N2 at 273–295 K and 1.0 bar, and a large heat of CO2 adsorption can be achieved at zero coverage (20.37 kJ mol−1for AlOF-1, 18.31 kJ mol−1 for GaOF-1 and 11.98 kJ mol−1 for InOF-1).
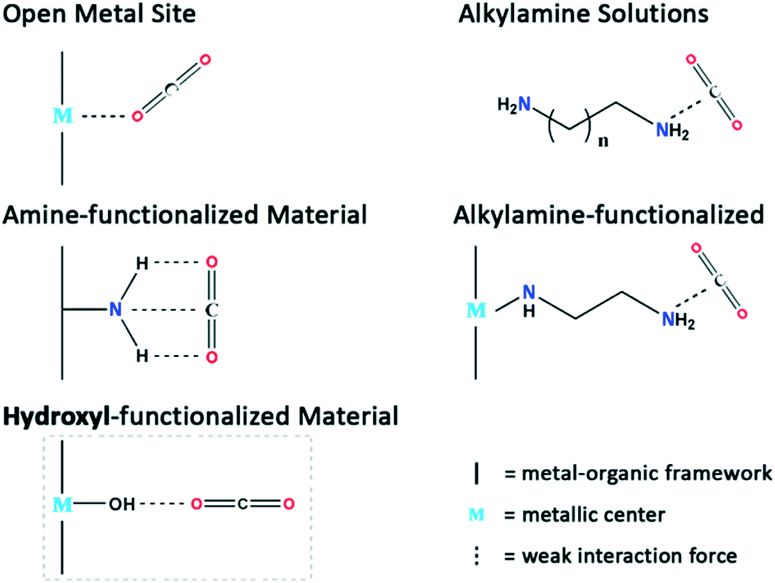 | ||
| Fig. 1 CO2 binding interactions in various MOF materials with different functional groups. In an OMS system, the CO2 molecule uses its central carbon to connect to the MOF metallic center. The alkylamine solution, NH2-functionalized, and alkylamine-functionalized MOF generally capture CO2 molecules through a side-on mode, whereas in the OH-functionalized system, the CO2 molecule attacks the OH group through an end-on mode. | ||
The pristine InOF-1 and ethanol (EtOH)-impregnated InOF-1 (InOF-1–EtOH) simultaneously exhibit an outstanding CO2 capacity under the same conditions.6 Therefore, it is of great importance to understand the in-depth mechanism for the binding formation of small molecules and their hosts using direct visualization of the interaction between the CO2 molecules and the InOF-1 framework. In this paper, the adsorption of CO2 in one water-stable tetracarboxylate-based framework with an In(III) metal cation, [InOF-1, Fig. 2a and b and S2 (ESI†)] is mainly predicted and explained using DFT calculations. Meanwhile, the electron and energy properties of InOF-1 were calculated using a combination of DFT and plane-wave pseudopotential methods as implemented in the Vienna Ab initio Simulation Package (VASP) code.8 Calculations were performed under the Perdew–Burke–Ernzerhof (PBE) approximation9 for exchange and correlation with a plane-wave cutoff energy of 380 eV. The optimized unit cell used has 72 and 84 atoms for the bare and four CO2 molecules loaded materials (InOF-1·4CO2, where each μ2-OH group only connects to one CO2 molecule), respectively. The wave functions were sampled according to the Monkhorst–Pack scheme with a k-point with mesh spacing of ∼0.05 Å−1.
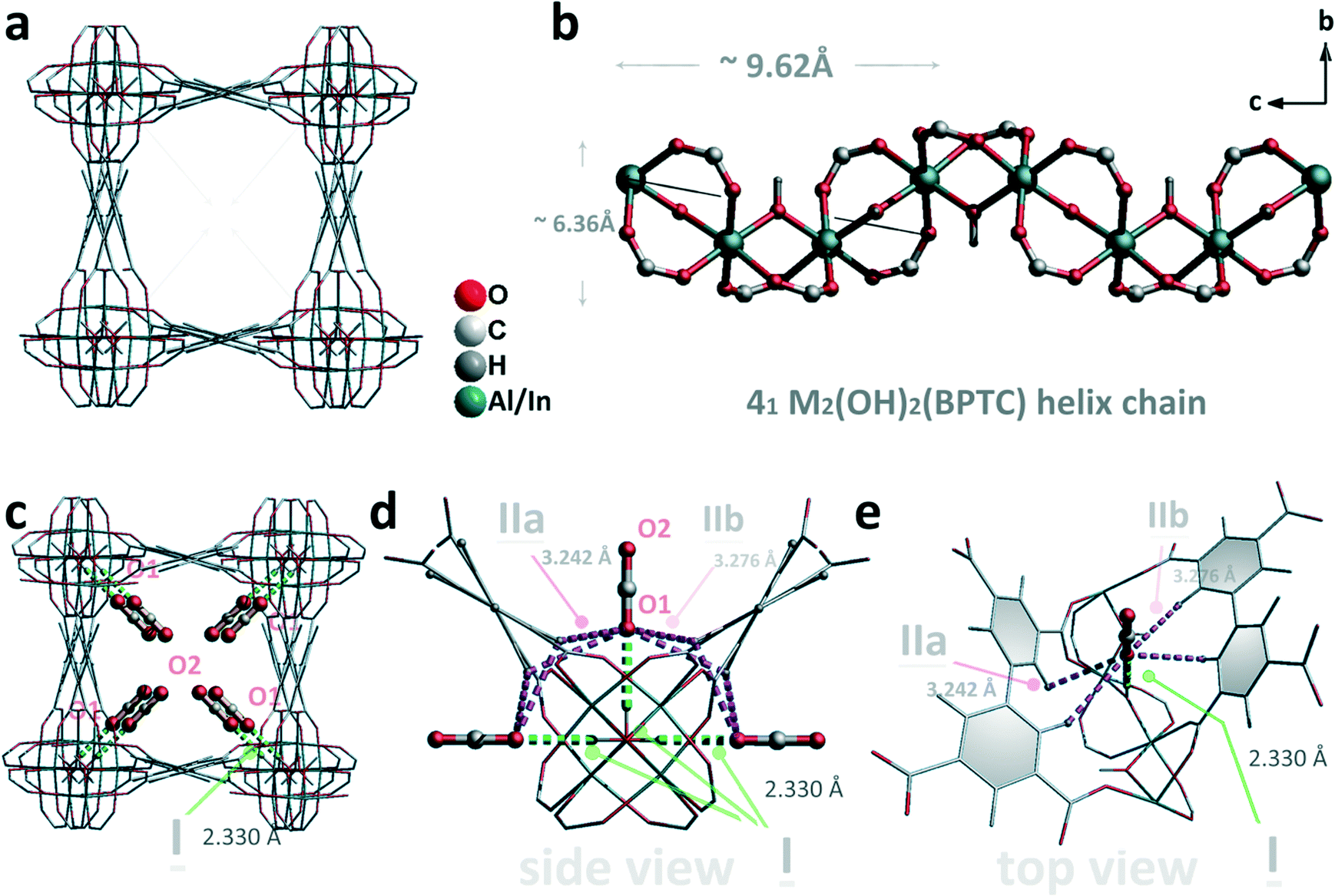 | ||
| Fig. 2 Theoretically optimized CO2 positions in the tetragonal channels of two isostructural M-BPTC frameworks. (a) View of the 3-dimensional structure with a square channel. The cis-μ2-OH groups protrude into the center of the channel from four directions. (b) View of the corner sharing octahedral [M2(OH)2(BPTC)] chain along the a axis. The different sizes of the 41 chain are highlighted. (c) View of the structure of M-BPTC·4CO2 obtained using a combination of DFT and plane-wave pseudopotential methods as implemented in the VASP software. The adsorbed CO2 molecules in the channel are highlighted by the use of the ball-and-stick mode. The interactions between CO2 molecules and μ2-OH groups are highlighted in green. (d and e) Detailed views of the roles of the –OH and –CH groups in binding CO2 molecules in a pocket-like cavity. | ||
As well as the InOF-1 material, two other isostructural host AlOF-1 and GaOF-1 were also prepared,7d–f and these possess a hydroxyl-functionalized 41 M2(OH2)(BPTC) helix chain (M = Al or Ga), whose sizes are ∼6.36 × 9.62 Å2 (Fig. 2b). DFT calculations were used to refine the crystal structure for the optimized InOF-1·4CO2 structures, and this shows that these adsorbed CO2 molecules interact with the cis-μ2-OH groups in an end-on mode (Fig. 2c). The O⋯H distance (dI) between the entrapped CO2 molecule and the hydroxyl group (HOH) is 2.330 Å which indicates a relatively moderate to weak hydrogen bond (Table 1). The optimized C–O bond lengths in CO2 were 1.179 Å (hydrogen bonded end, C–O1) and 1.173 Å (free end, C–O2), and the ∠OCO bond angle was absolutely linear and had a value of 180°. For the coordination environment of each captured CO2 molecule, the hydrogen bonded O1 atom was also bolstered together by weak cooperative supramolecular interactions between the O1 atom and H atoms from biphenyl C–H groups (HCH, O⋯H = 3.242 Å and 3.276 Å for dIIa and dIIb, respectively, and each occurs twice, Fig. 2d). In this context, the dIIa and dIIb distances in the InOF-1·4CO2 and GaOF-1·4CO2 structure (dIIb = 3.202 Å) are slightly larger than that of the AlOF-1·4CO2 structure (dIIa = 3.124 Å, dIIb = 3.197 Å, see Table 1), which obviously indicates more strong interactions between the main MOF framework and the captured CO2 molecules in the AlOF-1 parent. A top view of the adsorbed CO2 molecule in the OH-functionalized in-based chain, a total of 5 H atoms (4 HCH atoms and 1 HOH atom) attract cooperatively with the O1 charge centres of the captured CO2 molecules in the tetragonal channel through the combination of the moderate to weak hydrogen bonds and the supramolecular interactions (Fig. 2e). As shown in Fig. 2c–e, the modest hydrogen bond between the O1 atom and the HOH atom from the M–OH moiety is highlighted in green, and the weak cooperative hydrogen-bond interactions between O1 and HCH H(dp) from the C–H groups are highlighted in pink for clarity and comparison.
| Items | AlOF-1·4CO2 | GaOF-1·4CO2 | InOF-1·4CO2 |
|---|---|---|---|
| d C–O1 (Å) | 1.178 | 1.176 | 1.179 |
| d C–O2 (Å) | 1.174 | 1.170 | 1.173 |
| d I (Å) | 2.487 | 2.289 | 2.330 |
| d IIa (Å) | 3.124 | 3.086 | 3.242 |
| d IIb (Å) | 3.197 | 3.202 | 3.276 |
| ΔE (kJ mol−1) | 17.078 | 15.970 | 15.953 |
| ZPEC (kJ mol−1) | 9.190 | 8.069 | 6.068 |
| TEC (kJ mol−1) | 35.827 | 32.663 | 27.077 |
| ΔH (kJ mol−1) | 78.341 | 65.568 | 60.215 |
However, in order to reveal the theory of the adsorption of CO2 in these three types of isostructures, the binding energy (ΔE), zero point energy correction (ZPEC), thermal energy correction (TEC) and binding enthalpy (ΔH) of AlOF-1·4CO2, GaOF-1·4CO2 and InOF-1·4CO2 were calculated.
The ΔE of CO2 is evaluated using the following equation:
| ΔE = −EMOF·4CO2 + EMOF + 4ECO2 | (1) |
The ZPEC for a system is then calculated as:10
| ZPEC = ZPEMOF·4CO2 − ZPEMOF − 4ZPEBCO2 | (2) |
Similar to ZPEC, TEC is also t calculated as follows:10
| TEC = TEMOF·4CO2(T) − TEMOF(T) − 4TECO2(T) | (3) |
The binding enthalpies at a given temperature are calculated as:10
| −ΔH(T) = H(T)MOF·4CO2 − H(T)MOF − 4H(T)CO2 | (4) |
The previously described calculated energies are summarized in Table 1. In this table, the larger ΔE and ΔH represent stronger binding. Larger ZPEC and TEC values represent greater correction, that is to say, AlOF-1 (ZPEC = 9.910 kJ mol−1, TEC = 35.827 kJ mol−1) tends to possess greater correction with CO2 than both GaOF-1 and InOF-1. A stronger binding generates greater confinement and a steeper potential well, and thus yields a larger ZPEC but less thermal motion inside the well.10 This means that AlOF-1·4CO2 is easier to form than GaOF-1·4CO2 or InOF-1·4CO2. Consequently, agreement is achieved between the previously reported results and this data.
To compare the previously calculated results with experimental CO2 adsorption tests, InOF-1, AlOF-1 as well as GaOF-1 materials were synthesized according to previous methods reported in the literature for better comparison.5a,7d,f,11 In order to confirm the permanent porosity, the N2 isotherms at 77 K were first determined prior to the CO2 sorption test. The desolvated InOF-1 and GaOF-1 samples showed totally reversible type-I isotherms with the maximum uptake value of 270.9/131.1 m3 g−1 at 1.0 bar and 77 K which corresponds to a Brunauer–Emmett–Teller (BET) and Langmuir surface area of 1065/1093 m2 g−1 and 517/570 m2 g−1, respectively. In contrast, the activated AlOF-1 powder exhibited barely any N2 sorption capacity at 77 K, with a maximum value of only 13.4 cm3 g−1 at 1.0 bar and 77 K (Fig. 3a). Despite the microporous window size and the rigid framework for AlOF-1, it was surprising that the low N2 diffusion into the voids can be observed at 77 K. It was assumed this behaviour might be derived from a strong interaction between the nitrogen molecules and the narrow pore windows by the considerable quadrupole interactions with the electrostatic field gradients near the surface, which subsequently prohibit other molecules from penetrating into the voids,12 because the AlOF-1 framework possesses only open 1-dimensional channels along the c-axis.
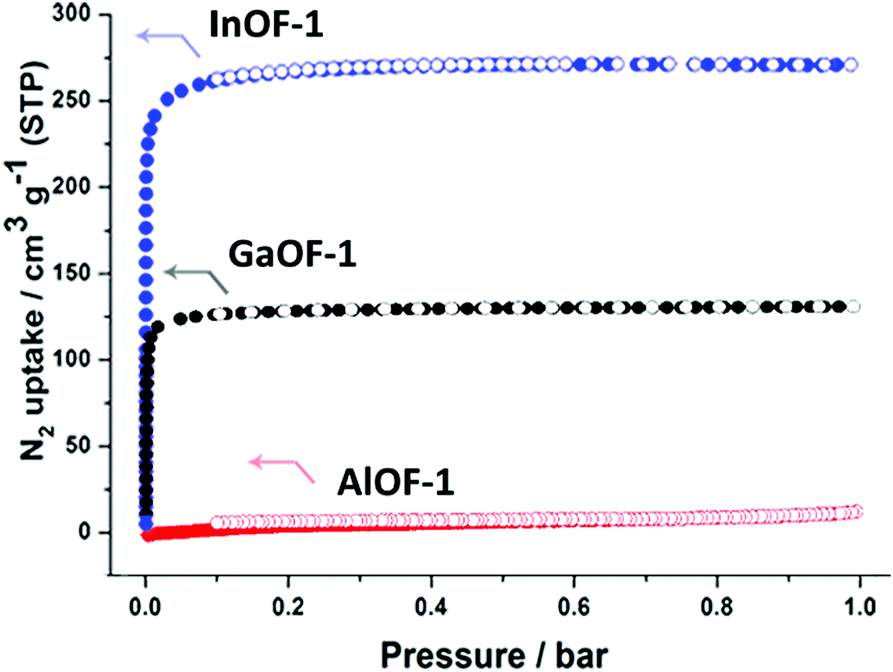 | ||
| Fig. 3 N2 adsorption/desorption isotherms at 77 K for AlOF-1, GaOF-1 and InOF-1. ● adsorption, ○ desorption. | ||
Most importantly, the fascinating internal OH-suspended tetragonal tubes inevitably prompt the further investigation of the practical CO2 sorption capacity. Single component low-pressure gas sorption isotherms for the three types of desolvated samples toward CO2 at 273 K, 283 K, and 295 K were collected using a volumetric measurement method and the results obtained are presented in Fig. 4a and Fig. S9–11 (ESI†). Compared to its very low sorption for N2, the CO2 isotherms of AlOF-1 at the specific temperatures (273–295 K) show extremely high sorption capacities, with the saturated value of 155.5 cm3 g−1 (6.94 mmol g−1, 305.4 mg g−1) at 273 K and 1.0 bar. Meanwhile, it was also found that the CO2 adsorption curve quickly reached the value of 42.6 cm3 g−1 (1.90 mmol g−1, 83.7 mg g−1) at 0.15 bar, which is lower than the equivalent partial pressure in flue gas (Fig. 4a). Obviously, this capacity surpasses the InOF-1 adsorption value of 39.1 cm3 g−1 (1.75 mmol g−1, 76.8 mg g−1) at 273 K and 0.15 bar and 140.1 cm3 g−1 (6.25 mmol g−1, 275.2 mg g−1) at 273 K and 1.0 bar. Similar trends can also be found in the uptake of CO2 at 1.0 bar and 283 K and 295 K, where the AlOF-1 shows a good performance with 118.6 cm3 g−1 and 86.7 cm3 g−1, respectively, whereas the InOF-1 exhibits a lower capability towards CO2 with 109.5 cm3 g−1 at 283 K and 83.9 cm3 g−1 at 295 K. Furthermore, in terms of CO2 storage, the Ga-based material shows the lowest capability of 90.3, 71.6 and 56.6 cm3 g−1 [Fig. 4b and S15 (ESI†)]. In this case, it was speculated that there exists a strong interaction between the solvent molecules and the GaOF-1 framework, thus leading to the incomplete desolvation and inadequate utilization of the expected pore volume. However, it should be noted that the CO2 capacity of AlOF-1 and InOF-1 materials gradually gets closer when the temperature is elevated from 273 K to 295 K (Fig. 4a), which means that the active CO2 spacing in AlOF-1 is lost faster than that in InOF-1. However, a large amount of CO2 is still retained under these different temperatures, especially for the Al-based framework, which indicates the excellent retention of the microporous nature before and after the desolvation treatment and gas sorption, as shown in Fig. S6 and S7 (ESI†).
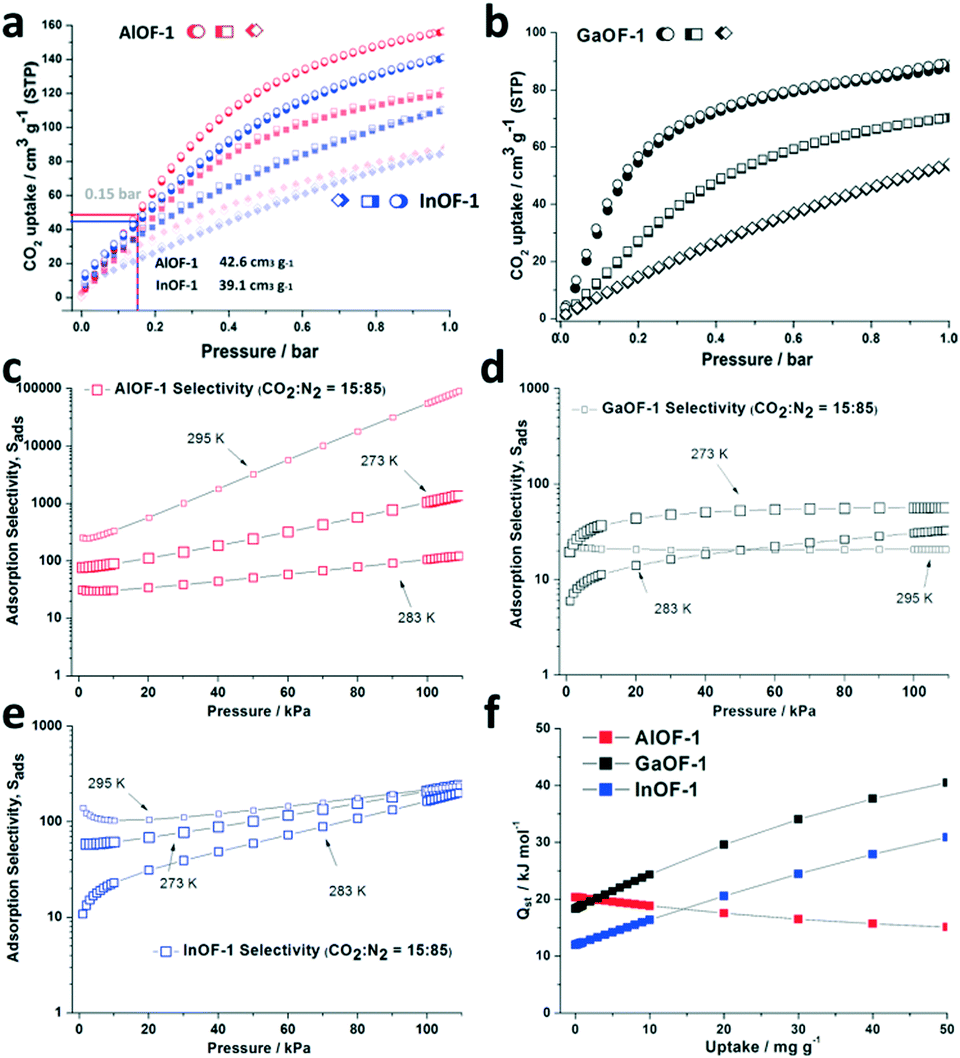 | ||
| Fig. 4 (a) and (b) Experimental CO2 sorption isotherms at 273 K (● adsorption, ○ desorption), 283 K (■ adsorption, □ desorption) and 295 K (◆ adsorption, ◇ desorption). (c)–(e) The selectivity between CO2 and N2 at three different temperatures for AlOF-1, GaOF-1 and InOF-1. (f) The adsorption heat (Qst) of CO2 for the three types of isostructural M-BPTC materials fitted using the virial method. | ||
Finally, the IAST prediction based upon the experimental CO2 and N2 isotherms are clearly presented in Fig. 4b. The adsorption selectivity is defined as Si/j = (q1/q2)/(p1/p2), in which qi is the amount of i adsorbed and pi is the partial pressure of i in the mixture. At 1.0 bar, the calculated CO2/N2 selectivities for AlOF-1 are more variable: 1078.8 and 108.4 at 273 K and 283 K from gas–phase mixtures in a 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85 molar ratio, whereas at 295 K, it reaches an excessive amount of 58571.6 because of the very low capacity of N2 at 1.0 bar. Compared to that of the Al-based material, the theoretically calculated CO2/N2 selectivity values for GaOF-1 and InOF-1 are more reasonable and trustworthy, where they are 56.7/212.6, 31.1/168.1 and 20.8/219.6 at 273, 283 and 295 K, respectively, [Fig. 4c–e, S20–22 (ESI†)]. More interestingly, the heat of CO2 adsorption at zero coverage is calculated to be 20.37 kJ mol−1 (AlOF-1), 18.31 kJ mol−1 (GaOF-1), and 11.98 kJ mol−1 (InOF-1) based on the CO2 isotherms at 283 K and 295 K, which indicates a more reasonable order for this series of M-BPTC structures despite the deficient performance for GaOF-1 in the CO2 sorption analysis (Fig. 4f). These calculations and observation indicate that all the OH-functionalized materials are promising candidates for practical CO2 capture and conversion applications.
:85 molar ratio, whereas at 295 K, it reaches an excessive amount of 58571.6 because of the very low capacity of N2 at 1.0 bar. Compared to that of the Al-based material, the theoretically calculated CO2/N2 selectivity values for GaOF-1 and InOF-1 are more reasonable and trustworthy, where they are 56.7/212.6, 31.1/168.1 and 20.8/219.6 at 273, 283 and 295 K, respectively, [Fig. 4c–e, S20–22 (ESI†)]. More interestingly, the heat of CO2 adsorption at zero coverage is calculated to be 20.37 kJ mol−1 (AlOF-1), 18.31 kJ mol−1 (GaOF-1), and 11.98 kJ mol−1 (InOF-1) based on the CO2 isotherms at 283 K and 295 K, which indicates a more reasonable order for this series of M-BPTC structures despite the deficient performance for GaOF-1 in the CO2 sorption analysis (Fig. 4f). These calculations and observation indicate that all the OH-functionalized materials are promising candidates for practical CO2 capture and conversion applications.
In summary, with a combination of theoretical calculations and experimental tests, the role of group A(III) metals in three types of OH-appended M-BPTC MOFs, including AlOF-1, GaOF-1 and InOF-1 were thoroughly compared. In this series, it was found that the CO2 binding to the cis-μ2-OH groups is in an end-on mode and this is further supported by H-bonding with adjacent biphenyl rings. The experiments and IAST results demonstrate that the largest binding energy and adsorption heat exist in the Al(III)–OH⋯C(O)2 system which could be empirically predicted by the atomic mass of the cations in the isostructural frameworks. This research provides a useful guideline for the future development of versatile MOFs with functionalized groups.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (51420105002, 21601137, 51602226), and Natural Science Foundation of Zhejiang Province (LQ16B010003), Yunnan Applied Basic Research Projects (2016FD083) and Scientific Research Foundation of the Education Department of Yunnan Province (2016ZZX229).Notes and references
-
(a) A. Schoedel, Z. Ji and O. M. Yaghi, Nat. Energy, 2016, 1, 16034 CrossRef CAS
; (b) N. Kornienko, Y. B. Zhao, C. S. Kley, C. H. Zhu, D. Kim, S. Lin, C. J. Chang, O. M. Yaghi and P. D. Yang, J. Am. Chem. Soc., 2015, 137, 14129–14135 CrossRef CAS PubMed
- C. McGlade and P. Ekins, Nature, 2015, 517, 187–190 CrossRef CAS PubMed
-
(a) G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed
-
(a) Y. Yan, M. Juríček, F. Coudert, N. A. Vermeulen, S. Grunder, A. Dailly, W. Lewis, A. J. Blake, J. F. Stoddart and M. Schröder, J. Am. Chem. Soc., 2016, 138, 3371–3381 CrossRef CAS PubMed
-
(a) J. J. Qian, F. L. Jiang, D. Q. Yuan, M. Y. Wu, S. Q. Zhang, L. J. Zhang and M. C. Hong, Chem. Commun., 2012, 48, 9696–9698 RSC
- R. A. Peralta, A. Campos-Reales-Pineda, H. Pfeiffer, J. R. Álvarez, J. A. Zárate, J. Balmaseda, E. González-Zamor, A. Martínez, D. Martínez-Otero, V. Jancik and I. A. Ibarra, Chem. Commun., 2016, 52, 10273–10276 RSC
-
(a) W. Zhou, H. Wu and T. Yildirim, J. Am. Chem. Soc., 2008, 130, 15268–15269 CrossRef CAS PubMed
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
-
(a) K. Lee, J. D. Howe, L. C. Lin, B. Smit and J. B. Neaton, Chem. Mater., 2015, 27, 668–678 CrossRef CAS
- T. Panda, S. Horike, K. Hagi, N. Ogiwara, K. Kadota, T. Itakura, M. Tsujimoto and S. Kitagawa, Angew. Chem., 2017, 129, 2453–2457 CrossRef
-
(a) K. M. Ok, J. Sung, G. Hu, R. M. J. Jacobs and D. O'Hare, J. Am. Chem. Soc., 2008, 130, 3762–3763 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: For the optimized structures: Re_GaOF-1 (a = 12.1338, b = 12.1339, c = 12.1338, P1), Re_GaOF-1 + 4CO2 (a = 14.9392, b = 14.9392, c = 11.9401, I4122), Re_InOF-1 (unit cell parameters: a = 12.76990, b = 12.77034, c = 12.76990, P1), Re_InOF-1 + 4CO2 (a = 15.7375, b = 15.7375, c = 12.5286, I4122), Re_AlOF-1 (a = 12.13385, b = 12.13386, c = 12.13385, P1), Re_AlOF-1 + 4CO2 (a = 14.9392, b = 14.9392, c = 11.9401, I4122). For more details please see the data in the ESI. See DOI: 10.1039/c7ce01195d |
| This journal is © The Royal Society of Chemistry 2017 |