
Recent advances on supramolecular isomerism in metal organic frameworks
Anirban
Karmakar
 *,
Anup
Paul
and
Armando J. L.
Pombeiro
*
*,
Anup
Paul
and
Armando J. L.
Pombeiro
*
Centro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049–001, Lisbon, Portugal. E-mail: anirbanchem@gmail.com; pombeiro@tecnico.ulisboa.pt
First published on 7th July 2017
Abstract
Metal–organic frameworks (MOFs) or coordination polymers (CPs), a class of compounds constructed often by self-assembly of inorganic metal ions and organic ligands, have attracted growing interest on account of their diverse structures, permanent porosity and tunable properties, with a prospect for various applications. In some cases, the self-assembly of the building blocks produces different MOFs with the same molecular composition, i.e., supramolecular isomers. The design of supramolecular isomers in the area of MOFs is challenging and this highlight gathers recently published articles concerning the different types of supramolecular isomers, i.e., structural, conformational, catenane and optical, in this area. The syntheses and structures of the isomers and their comparison are presented, as well as the role of various parameters, such as temperature, solvent, concentration, crystallization method, additive and template, in the formation of the different supramolecular isomers, by analyzing selected examples.
 Anirban Karmakar | Anirban Karmakar received his M.Sc. degree from University of Burdwan, India in 2005. He earned his Ph.D. in Inorganic Chemistry under the supervision of Prof. Jubaraj Bikash Baruah in 2009 from the Indian Institute of Technology Guwahati, India. Afterwards, he worked as a postdoctoral research associate in the groups of the Prof. Israel Goldberg (Tel Aviv University, Israel) and Prof. Lars Öhrström (Chalmers University Of Technology, Sweden). Currently, he is a postdoctoral researcher in Prof. Armando J. L. Pombeiro's research group at Instituto Superior Técnico, Portugal. His research interests include design and synthesis of metal organic frameworks, heterogeneous catalysis, crystal engineering and host–guest chemistry. |
 Anup Paul | Anup Paul was born in Tura, Meghalaya, India. He obtained his M.Sc. in 2005 and Ph.D. in 2012 from North-Eastern Hill University (NEHU), Shillong, India under the supervision of Prof. T. S. Basu Baul. He worked with Prof. D. S. Pandey as a Dr. D. S. Kothari postdoctoral fellow in Banaras Hindu University, Varanasi, India for almost a year and then joined Instituto Superior Tecnico, Lisbon, Portugal as an FCT postdoctoral fellow under the supervision of Prof. A. J. L. Pombeiro. His current research interest is in bioinorganic chemistry and in the construction and application of porous coordination polymers in catalysis and sensing. |
 Armando J. L. Pombeiro | Armando J. L. Pombeiro is a Full Professor at Instituto Superior Técnico, Universidade de Lisboa. His research group investigates the activation of small molecules with industrial, environmental or biological significance, including metal-mediated synthesis and catalysis, crystal engineering of coordination compounds, design and self-assembly of polynuclear and supramolecular structures, molecular electrochemistry and theoretical studies. He authored one book, edited 4 books, (co-)authored over 600 research publications and 33 patents, and presented 100 invited lectures at international conferences. His work has received over 16 |
1. Introduction
The construction of new metal organic frameworks (MOFs) or coordination polymers (CPs) has been the subject of significant interest over recent decades as a new frontier for material research.1 This is due not only to their interesting architectures and topologies but also to their potential applications in various fields, such as heterogeneous catalysis,2 nonlinear optics,3 gas storage and separation,4 and magnetism.5 Nevertheless, the design and building of high-dimensional polymeric networks is still rather challenging.For the construction of suitable MOFs, the choice of ligands or metal ions and the reactivity and solubility of the linkers play an important role.6 However, many other factors, e.g., temperature, solvent, reaction pH, additive, template and auxiliary ligands, can affect the development and topology of the MOFs.7 Thus, their controlled synthesis and structure prediction are still very thought-provoking.
The self-assembly of a metal ion and a ligand can produce more than one MOF having different structures and properties. In some cases, the obtained different frameworks have the same chemical composition, being termed as “Supramolecular Isomers” and the phenomenon being designated by “Supramolecular Isomerism”. The term “Polymorphism” is also used to describe the different crystal forms of the same chemical species, often reflected by different crystal packings, space groups and unit cell parameters.8 However, MOFs often contain pores that are filled with solvent and these guest molecules can be different or their numbers associated with the frameworks can be distinct. Hence, we cannot describe them as polymorphs. The term “Polymorphism” appears to be more appropriate for molecular crystals rather than for polymeric networks.
Supramolecular isomerism in MOFs (or CPs) was first reviewed by Moulton and Zaworotko in 2001.9 Later, Chen et al. reported a review on supramolecular isomerism in MOFs based on N-donor ligands,10 and, in 2011, Zhou and co-workers published a review on this topic.11 Zaworotko et al. divided supramolecular isomerism into four main categories: structural, conformational, catenane or interpenetration and optical supramolecular isomerisms.9 The term structural supramolecular isomerism is used to describe the different superstructures with the same molecular composition of the network. The conformational and catenane supramolecular isomerisms arise from the different conformations of flexible ligands and the different degrees of interpenetration in the frameworks, respectively. When the frameworks with the same molecular components crystallize in different chiralities, they are termed as optical supramolecular isomers. Besides these, there are a few other types of isomers, such as orientation isomers [different orientations of the ligands or secondary building units (SBUs) within the crystal structures],11 topological isomers (similar local coordination environments but different topologies)12 and distortional supramolecular isomers (differ only in the length of one or more bonds due to crystallographic disorder).13 For such isomers, the different features of each isomer framework often require further studies to understand them.
Recently, many new supramolecular isomers of MOFs based on carboxylate14 and N-containing ligands15 have appeared, but their collection in a review format is missing. In this highlight, we aim to illustrate the main types of supramolecular isomers, namely structural, conformational, catenane and optical ones, in the area of MOFs with illustrative examples from the literature. Attention is also paid to the synthetic strategies and the crystal structures of individual isomers. Both similarities and differences in the structures and properties of each group are discussed. The role of various parameters, namely the temperature, solvent, concentration, crystallization method, additive and template, in the formation of different supramolecular isomers is also addressed by analyzing selected examples.
2. Structural supramolecular isomerism
The structural supramolecular isomerism of MOFs concerns the structural diversity of compounds having the same chemical composition. This isomerism arises mainly due to differences in the coordination modes of binding multidentate ligands. Several factors, such as temperature, solvent, pH, crystallization method or template, can be responsible for the construction of different structural supramolecular isomers.16 The following sections address some interesting cases.2.1 Solvent-dependent structural isomerism
An example of solvent-dependent structural isomerism in MOFs was reported19 by our group. We have synthesized three 1D zinc(II) CPs by the solvothermal reactions of zinc(II) nitrate hexahydrate and 5-acetamidoisophthalic acid (H2L1) in the presence of a dimethyl formamide and methanol mixture, formamide or N-methylformamide, leading to the formation of [Zn(L1)(H2O)2]n (1), [Zn(L1)(H2O)2]n·nFA (2; FA = formamide) or [Zn(L1)(H2O)2]n·nNMF (3; NMF = N-methylformamide), respectively (L = 5-acetamidoisophthalate).The single crystal X-ray diffraction analysis revealed that 1 exhibits a left-handed helical 1D structure, whereas 2 and 3 have zigzag-type 1D chains (Fig. 1). These compounds have identical asymmetric units except for the absence (in 1) or presence (in 2 and 3) of solvent molecules. The asymmetric units of these compounds contain one zinc(II) ion, one L2− ligand, two coordinated water molecules and one non-coordinated molecule of formamide (in 2) or N-methylformamide (in 3). The Zn2+ centres have tetrahedral geometries in 1 and 2 (τ4 = 0.93 and 0.83, respectively)17 and a trigonal bipyramidal geometry in 3 (τ5 = 0.54).18
 | ||
| Fig. 1 1D crystal structures of compounds 1 (helical), 2 (zig-zag) and 3 (zig-zag). Reprinted (adapted) with permission from ref. 19. Copyright 2015 American Chemical Society. | ||
In these polymers, the structural difference mainly arises from the coplanarity or non-coplanarity of the carboxylate groups with respect to the phenyl ring of the ligand. For 1, one carboxylate group lies on the plane of the phenyl ring of the ligand, but the other one is twisted and forms a dihedral angle of 54.70° with the phenyl ring of the ligand, accounting for the helical nature of this framework. A similar phenomenon was observed for compound 3, the dihedral angle between the carboxylate group and the aromatic ring being ca. 13.02°. However, for compound 2, both carboxylate groups are co-planar with the phenyl ring of the ligand (dihedral angle = 0.0°) which determines the flat nature of this 1D polymer.19
Another example of structural isomers based on the amidoisophthalic acid ligand was also described20 by our group. We have synthesized the 1D zinc-based CPs [Zn(L2)(H2O)2]n (4) and [Zn(L2)(H2O)2]n·2nH2O (5) by the solvothermal reaction of 5-benzamidoisophthalic acid (H2L2) with zinc(II) nitrate hexahydrate in the presence of N,N-dimethylformamide (DMF) and methanol (MeOH) (for 4) or DMF and 1,4-dioxane mixtures (for 5) (Fig. 2).
 | ||
| Fig. 2 Crystal structures of 1D frameworks of 4 (helical) and 5 (zig-zag). Reprinted (adapted) with permission from ref. 20. Copyright 2014 Royal Society of Chemistry. | ||
MOFs 4 and 5 have identical molecular formulae, except for the additional crystallization water molecules of the latter. Their structural analysis revealed that they have a helical and a zigzag-type one-dimensional structure, respectively, and they crystallize in different space groups (4 in P212121 and 5 in P![[1 with combining overline]](https://www.rsc.org/images/entities/char_0031_0305.gif) ). These isomeric compounds are also termed solvatomorphs.
). These isomeric compounds are also termed solvatomorphs.
Both CPs have identical asymmetric units which contain one Zn(II) ion, one deprotonated ligand (L22−) and two coordinated water molecules, but for 5 two uncoordinated water molecules are present. These MOFs display a tetrahedral coordination (τ4 = 0.92 for 4 and 0.89 for 5).17 For compound 4, the carboxylate groups are coordinated in an anti-fashion, whereas for 5, they are coordinated in a syn mode. This fact may be the main reason behind the different arrangements (helical and zig-zag) of the structures. This also disturbs the relative orientation of the phenyl groups of L22− and the least-square planes of adjacent ligands in 4 make an angle of 52.55° while in 5 that angle is nil. The topological analysis of 4 and 5 indicates that they can be represented as 2-connected uninodal nets with topological type 2C1. These CPs act as efficient heterogeneous catalysts for the diastereoselective nitroaldol (Henry) reaction of aldehydes with nitroalkanes and can be recycled without losing activity.20
Du et al. reported a pair of polymorphic structural isomers having the same molecular formula [Cd(L3)2]n (6 and 7) (L3 = 5-fluoronicotinate), synthesised from fluoronicotinic acid under different solvent conditions. Compound 6 was prepared by layering a CH3CN solution of the ligand onto a water solution of the Cd(II) salt. Upon adopting the same methodology and changing the reaction solvent to CH3OH–H2O, another 3D CP (7) was obtained.21 The single-crystal X-ray diffraction study revealed that both MOFs possess a 3D coordination network but with different topological structures (Fig. 3). The asymmetric unit of 6 contains one Cd(II) ion and two L3 ligands, but for 7 the asymmetric unit consists of one-half Cd(II) ion and one crystallographically independent L3 ligand. In both MOFs, the Cd(II) centres are octahedrally occupied by four carboxylate O and two pyridyl N donors from five (for 6) and six (for 7) ligands. For both frameworks, the dinuclear {Cd2(COO)2} moiety acts as a SBU with Cd⋯Cd distances of 4.128(1) Å (for 6) and 5.039(1) Å (for 7). Topological analysis revealed that framework 6 has a (3,5)-connected (4·62)(4·67·82) network, whereas 7 has a (3,6)-connected ant topology with the point symbol of (4·62)2(44·62·88·10).
 | ||
| Fig. 3 Synthesis and crystal structures of frameworks 6 and 7. Reprinted with permission from ref. 21. Copyright 2013 Royal Society of Chemistry. | ||
Banerjee et al. reported22 two In(III)-based isomeric MOFs, [(CH3)2NH2]n[In(L4)2]n·2nH2O (8) and [(CH3)2NH2]n[In(L4)2]n·nDMF (9) (L4 = isophthalate), formed under slightly different solvothermal conditions (Fig. 4). Framework 8 was synthesised by the reaction of In(NO3)3·xH2O, isophthalic acid and tetramethyl ammonium chloride in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of DMF and H2O, at 120 °C for 96 h. Framework 9 was produced in the same way but using a 2:1 mixture of DMF and H2O. Single crystal structural analysis revealed that compound 8 crystallized in the orthorhombic Pna21 space group, whereas 9 crystallized in the monoclinic P21/c space group. For both compounds, each In(III) centre is occupied via seven carboxylate oxygen atoms from four different ligands. MOF 8 contains non-coordinated H2O and [(CH3)2NH2]+ cations, while 9 contains [(CH3)2NH2]+ cations and solvent (DMF) molecules inside the framework. In both MOFs, {In(COO)4} SBUs are connected via a L4 ligand forming 1D structures which are further interconnected by another L5 ligand generating 2D layer-type structures along the crystallographic ab plane (for 8) and bc plane (in 9).
:1 mixture of DMF and H2O, at 120 °C for 96 h. Framework 9 was produced in the same way but using a 2:1 mixture of DMF and H2O. Single crystal structural analysis revealed that compound 8 crystallized in the orthorhombic Pna21 space group, whereas 9 crystallized in the monoclinic P21/c space group. For both compounds, each In(III) centre is occupied via seven carboxylate oxygen atoms from four different ligands. MOF 8 contains non-coordinated H2O and [(CH3)2NH2]+ cations, while 9 contains [(CH3)2NH2]+ cations and solvent (DMF) molecules inside the framework. In both MOFs, {In(COO)4} SBUs are connected via a L4 ligand forming 1D structures which are further interconnected by another L5 ligand generating 2D layer-type structures along the crystallographic ab plane (for 8) and bc plane (in 9).
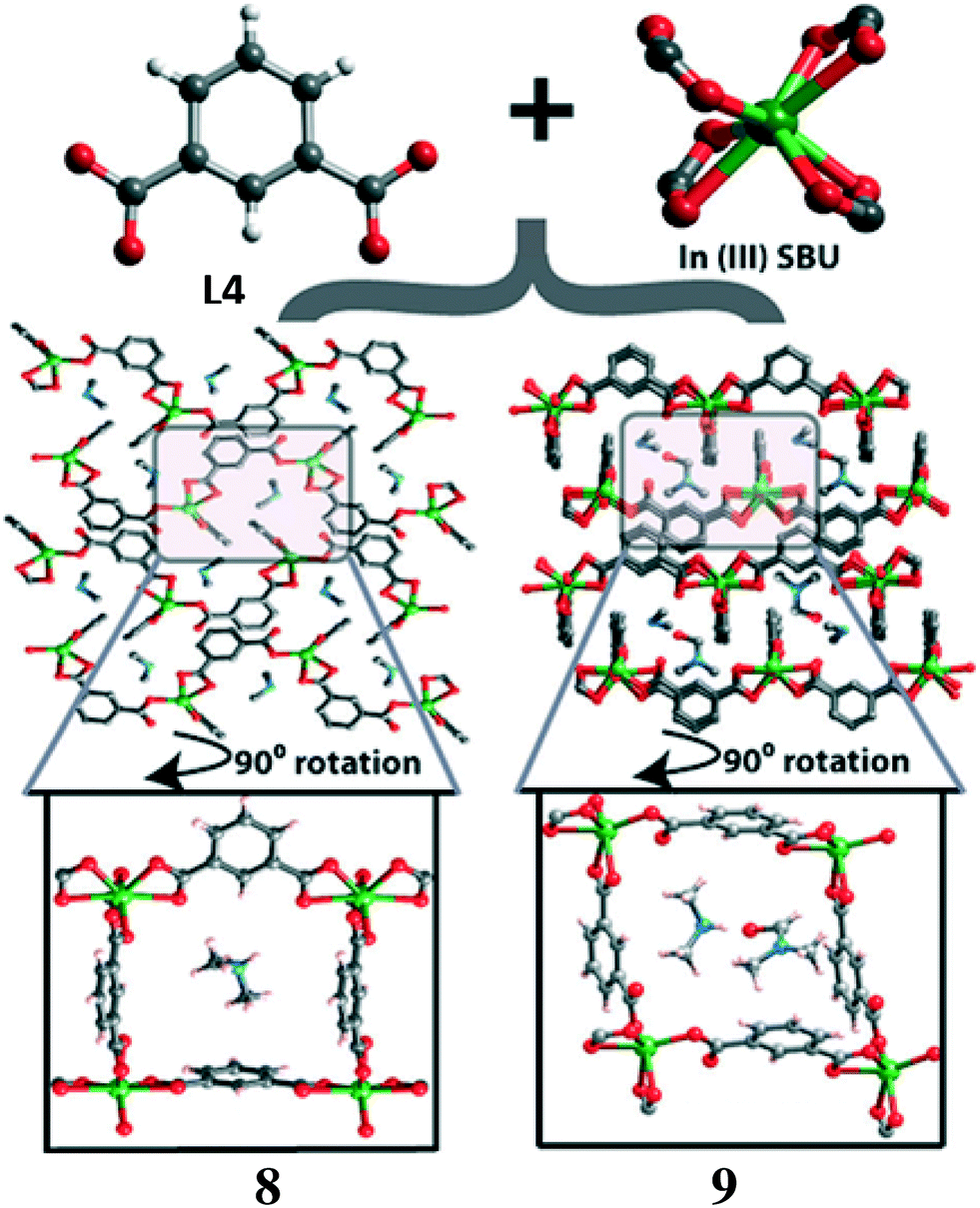 | ||
| Fig. 4 Schematic representations of the isophthalic acid, the {In(COO)4} SBU and the two dimensional structures of 8 and 9 (note: zoomed-in figures have been rotated 90° for clarity). Reprinted with permission from ref. 22. Copyright 2013 Royal Society of Chemistry. | ||
Compound 8 shows a higher proton conductivity (3.4 × 10−3 S cm−1) under humidified conditions (98% RH) than its isomer 9 (2.6 × 10−5 S cm−1). The greater proton conductivity shown by 8 can be accounted for by the presence of solvent H2O and [(CH3)2NH2]+ cations together, which may facilitate the proton conductivity under humidified conditions. Framework 9 contains DMF which may restrict the approachability of water molecules to [(CH3)2NH2]+ cations under humidified conditions, thus, showing a lower proton conductivity despite adopting a similar 2D network structure.
In another study, Lu et al. reported23 two new MOFs having the common formula [Cd(L5)(L6)2]n (10 and 11) [L5 = 2-amino-1,4-benzenedicarboxylate; L6 = methylenebis(3,5-dimethylpyrazole)], by tuning the composition of the reaction medium. The hydrothermal reaction of Cd(NO3)2·4H2O, H2L5 and L6 in water at 120 °C for 48 h led to the formation of 10, whereas 11 was synthesized under similar reaction conditions except that a 1:1 mixture of DMF and H2O was used instead of water (Fig. 5). Single-crystal diffraction analysis revealed that both frameworks possess 2D networks although they have crystallized in different space groups P2/c (for 10) and C2/c (for 11). Their asymmetric units consist of half Cd(II) ion, half L5 ligand and one L6 ligand. Each Cd(II) has a distorted octahedral geometry being coordinated by two O-atoms from two L5 ligands and four N-atoms from four different L6 ligands. However, there are subtle differences in some bond distances and angles between the two frameworks.
 | ||
| Fig. 5 Schematic representation of the synthesis of frameworks 10 and 11. Reprinted (adapted) with permission from ref. 23. Copyright 2013 Royal Society of Chemistry. | ||
In both 10 and 11, the Cd(II) centres are bridged by deprotonated L6 ligands to produce 1D infinite chains of {Cd(L5)}, which are further linked by cis-conformation L6 ligands generating 2D layer-type structures. Moreover, these 2D layers are assembled into 3D supramolecular networks with different voids (13.27 × 16.31 Å2 for 10 and 14.77 × 15.78 Å2 for 11). Solvent-accessible voids were calculated by PLATON analysis to be 8.1% (for 10) and 27.9% (for 11) of the unit cell volume.
Another example of genuine structural supramolecular isomers was reported by Huang and co-workers.24 They have synthesized two Co(II) MOFs with the common formula [Co(L7)(L8)]n (12 and 13), obtained by the hydro(solvo)thermal reaction of cobalt(II) acetate, 1,4-naphthalenedicarboxylic acid (H2L7) and 5,5′-dimethyl-2,2′-dipyridyl (L8) (Fig. 6). MOF 12 was obtained by carrying out the reaction at a temperature of 160 °C for 5 days using only H2O as solvent, while 13 could be obtained by carrying out the reaction using DMF as solvent. Interestingly, compound 13 could also be prepared by using another solvent such as MeOH, CH3CN, DMA, EtOH or a mixture of DMF and water, under the same reaction conditions. Moreover, it could also be synthesized within the wide temperature range of 130–190 °C, at an interval of 10 °C.
 | ||
| Fig. 6 (A) Synthetic scheme and crystal structures of the 1D double chain in 12 and the 2D layer in 13. The relative orientation of the L8 ligands in compounds 12 (B) and 13 (C). Reprinted (adapted) with permission from ref. 24. Copyright 2014 American Chemical Society. | ||
Single-crystal X-ray diffraction analyses revealed that 12 and 13 are supramolecular isomers which have crystallized in the triclinic P and orthorhombic Pbca space groups, respectively. Both compounds have the same asymmetric unit which contains one Co(II) ion, one L7 and one L8 ligand and has no solvent molecule present in the lattice. For both compounds, the distorted octahedral coordination geometry of each Co(II) centre is fulfilled by four O atoms from three L7 ligands and two N atoms from one L8 ligand. In these MOFs, the {Co2(OOCR)2} dimeric unit acts as a SBU, being interconnected through a pair of bridging L7 ligands giving rise to a 1D ladder-like chain of framework 12. However, for 13, two {Co2(OOCR)2} SBUs are connected only by one L7 bridging ligand forming a 2D sheet in the ab plane (Fig. 6).
The relative orientation of the L7 ligands is the main reason behind the major structural difference between both frameworks. For 12, both L7 ligands are parallel to each other, whereas for 13, only two of the three sets of naphthalene rings are parallel to each other while the remaining one forms a dihedral angle of 50.22(4)° with each of them. The orientational changes of the L7 ligands are the key factor in the formation of the isomers with different structural dimensionalities. These frameworks also display an interesting magnetic behaviour at low temperature. They show different magnetic interactions although they have the same {Co2(OOCR)2} dinuclear unit. The relative orientation of the L7 ligands may affect the orthogonality of the magnetic orbitals and may be responsible for the change from antiferromagnetic to ferromagnetic interaction between adjacent Co(II) ions at low temperature. Supramolecular interactions, such as hydrogen bond contacts or π–π stacking interactions, can also result in magnetic variations.
Apart from genuine supramolecular isomers, there are other varieties of supramolecular isomers where the basic framework remains the same but varies with the presence of solvent molecules in the crystal structure. Examples are discussed below.
Chen et al. reported25 three one-dimensional (1D) pseudopolymorphs of silver(I) 2-methylimidazolate, namely [Ag4(L9)4]n·n(C6H6) (14), [Ag4(L9)4]n·n(C8H10) (15; C8H10 = p-xylene or ethylbenzene) and [Ag(L9)]n (16) (HL9 = 2-methylimidazole). These CPs are structural supramolecular isomers containing different solvent molecules. They were obtained by carrying out the reactions in different solvents resulting in solvent-induced supramolecular isomers. The crystals of compound 14 were obtained by layering a solution of HL9 in methanol/benzene onto a solution of AgNO3 in aqueous ammonia. Compounds 15 and 16 were synthesized by using a similar methodology except that a mixture of methanol/p-xylene/ethylbenzene was used for synthesis of 15 and only methanol was used for 16.
The single crystal structural analysis showed that these polymers have different structures (Fig. 7). Compound 14 exhibits an unusual pseudo-81 helical structure, whereas 15 and 16 show S-shaped and zig-zag chain-type 1D structures, respectively. All these compounds have the same coordination environment around the Ag(I) ions but they have crystallized in different space groups, i.e., Pnna (for 14), P21/c (for 15) and P21/n (for 16). The asymmetric units of polymers 14 and 15 contain four crystallographically independent Ag(I) atoms and four L9 ligands, but in the case of 16 it comprises one crystallographically independent Ag(I) atom and one L9 ligand.
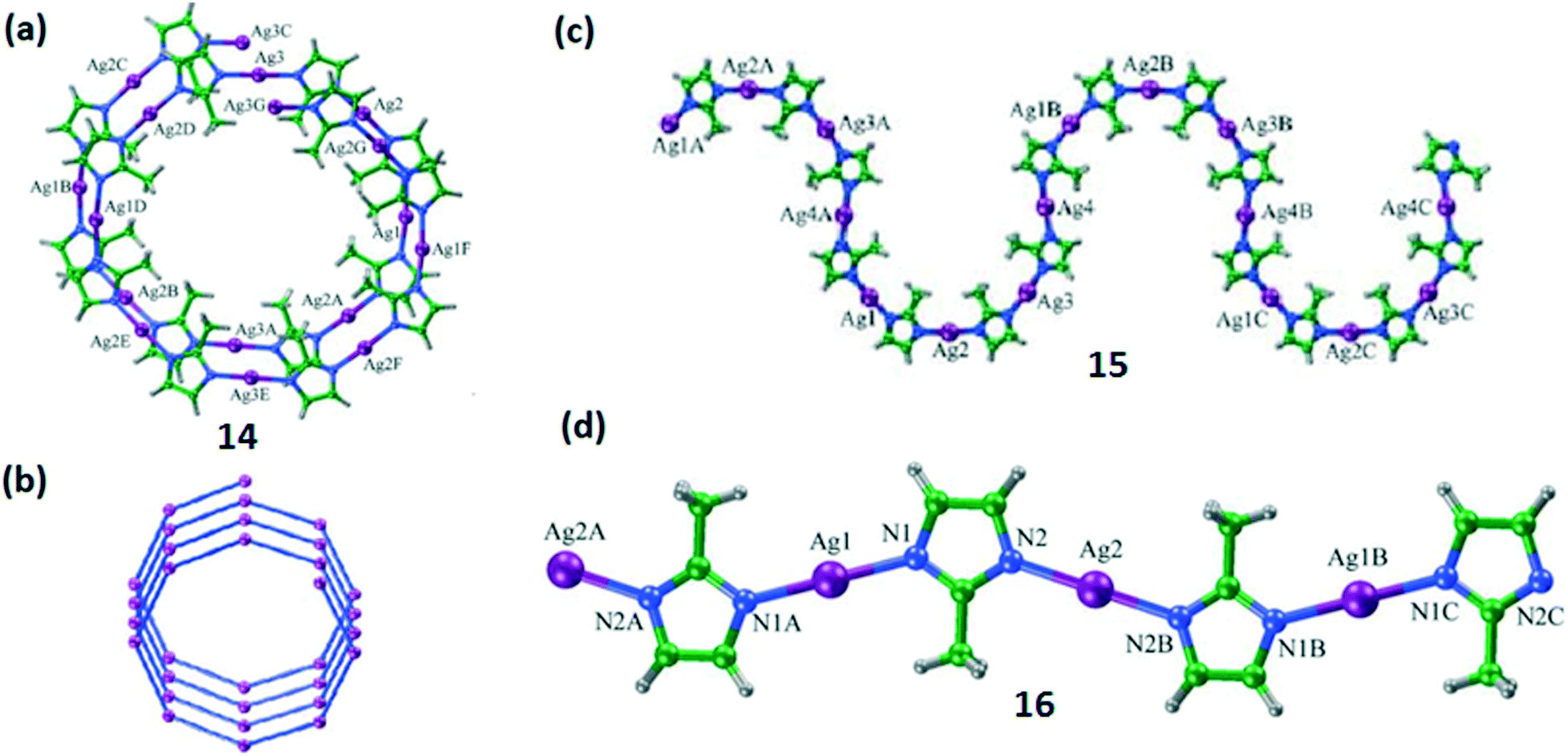 | ||
| Fig. 7 (a) and (b) Top view of the right-handed pseudo-81 helix in 14 along the b-axis. (c) 1-D S-shaped chain of 15 viewed along the c-axis. (d) Crystal structure of the 1-D zig-zag chain in 16. Reprinted (adapted) with permission from ref. 25. Copyright 2006 Royal Society of Chemistry. | ||
For compound 14, the ligands L9 connected to the Ag(I) ion are oriented in a syn-fashion with the dihedral angles in the range of 13.9–34.7°. Due to such a twisted orientation, a helical structure with all of the methyl groups in the L9 ligands pointing towards the channel is formed. For 15, the ligand L9 also coordinates in a syn-fashion and constructs an arc-shaped Ag4(L9)4 unit, which is further interconnected into an S-shaped one-dimensional chain. But in the case of 16, the ligand is coordinated in an anti-fashion which leads to a zig-zag type of structure.
Another example was reported26 by our group. We have synthesized five pseudo-polymorphic 1D MOFs, [Cu2(L10)2(bipy)]n·2nDMSO (17; DMSO = dimethyl sulfoxide), [Cu2(L10)2(bipy)]n·3nFA·nH2O (18), [Cu2(L10)2(bipy)]n·2nNMF (19), [Cu2(L10)2(bipy)]n·2nDMF (20) and [Cu2(L10)2(bipy)]n·2nMeOH (21) (bipy = 4,4′-bipyridine), derived from the acyclic Schiff base o-[(o-hydroxyphenyl)methylideneamino]benzenesulfonic acid (H2L10) in the presence of bipy. These CPs were obtained by the reaction of Cu(OAc)2·H2O and H2L10 in different solvents. For the synthesis of compounds 17–21, we have used dimethylsulfoxide, formamide, N-methylformamide, N,N-dimethylformamide and methanol, respectively (Scheme 1).
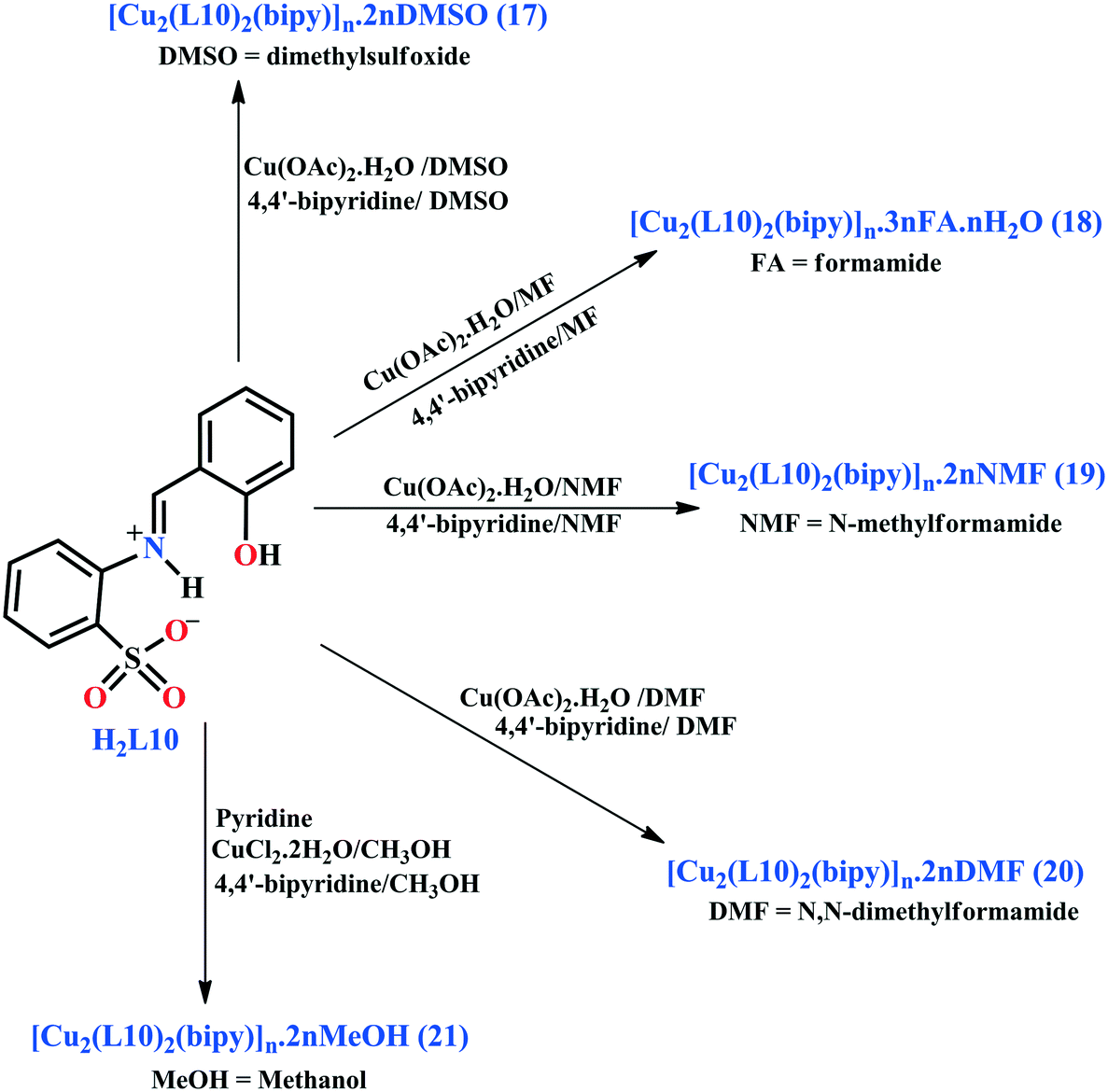 | ||
| Scheme 1 Syntheses of compounds 17–21 from the Schiff base H2L10. Reprinted (adapted) with permission from ref. 26. Copyright 2015 Royal Society of Chemistry. | ||
Crystal structure analyses revealed that all the compounds contain similar hacksaw-type 1D chains with variable non-coordinated solvent molecules (dimethylsulfoxide in 17, formamide and water in 18, N-methylformamide in 19, N,N-dimethylformamide in 20 and methanol in 21). The asymmetric units of these frameworks contain two Cu(II) ions, two L10 ligands and one 4,4′-bipyridine, and each Cu(II) ion adopts a slightly distorted square pyramidal geometry. In these compounds, the phenoxo oxygen is coordinated to the two Cu(II) ions and constructs {Cu2(Ophenoxo)2} cores which are further bridged by 4,4′-bipyridine and thus giving rise to 1D hacksaw chains.28
MOFs 17–20 are reversibly interconvertible with each other, but the insolubility in methanol prevents their conversion into the methanol-containing polymer 21. This provides a rare example of solvatomorphism in a metal–organic polymeric compound.
2.2 Temperature-dependent structural isomerism
Maji and co-workers reported27 four different Ni(II)-based MOFs, [Ni(bipy)(H2O)4]n(L11)n·4nH2O (22), [Ni(bipy)(H2O)4]n(L11)n·2nH2O (23) and [Ni(bipy)(H2O)4]n(L11)n (24 and 25), constructed from L11 (2,6-naphthalenedisulphonate) and bipy (4,4′-bipyridine). Although compounds 22 and 23 can also be regarded as supramolecular isomers, the typical concept of supramolecular isomers is followed only by compounds 24 and 25 (see below). These MOFs were synthesised from the same stoichiometric ratio of reagents but under different reaction temperatures. Framework 22 was synthesized at room temperature in H2O/EtOH medium, but 23, 24 and 25 were isolated under hydrothermal conditions at 140 °C, 120 °C and 100 °C, respectively (Fig. 8). | ||
| Fig. 8 Synthesis and packing diagrams of 24 and 25. Reprinted (adapted) with permission from ref. 27. Copyright 2009 American Chemical Society. | ||
The crystal structure analysis revealed that all the compounds contain the common one-dimensional building block [Ni(bipy)(H2O)4]2+ and the counter-ion L11. They have the same asymmetric unit [with one Ni(II) ion, one 4,4′-bipyridine and four coordinated water molecules] but crystallize in different space groups. They differ in the number of water molecules of crystallization and in the arrangement of the counter-ion L11. Compounds 24 and 25 are particularly interesting since they have the same molecular formula [Ni(bipy)(H2O)4]n(L11)n but exhibit different supramolecular arrangements. Thus, they can be designated as polymorphs, as well as structural supramolecular isomers. In all the compounds, the 1D chains of Ni–bipy are interconnected via strong hydrogen-bonding and π–π interactions, resulting in 3D supramolecular architectures. The structural differences of 24 and 25 mainly arise from the different orientations of the 1D metal–bipyridine chains and of the counter-ion L11, and the temperature plays an important role in their construction.
Two temperature-dependent genuine supramolecular isomers [Cu2(μ3-SCN)(μ4-L12)]n (26) and [Cu2(μ4-SCN)(μ4-L12)]n (27) (L12 = 5-methyl tetrazolate) were reported by Mahmoudi et al.28 Framework 26 was synthesized by the hydrothermal reaction of NaN3 and KSCN in acetonitrile, in the presence of [Cu(CH3CN)4]ClO4 at 135 °C for 72 h. On the other hand, compound 27 was obtained by performing the reaction under similar conditions but at a higher temperature (168 °C). The single crystal X-ray structural analysis revealed that compounds 26 and 27 have 2D and 3D structures, respectively; however, they crystallize in the same orthorhombic Pbam system. For both frameworks, the Cu(II) centre is tetrahedrally coordinated by two N atoms from two L12 ligands and two S atoms from two thiocyanate ligands. In both MOFs, the [Cu2(L14)]+n unit acts as a SBU, being interconnected by thiocyanate anions via μ3 and/or μ4 fashions. These compounds provide an example of structural supramolecular polymorphs.
Cai et al. reported29 two copper(II)-based supramolecular isomers 28 and 29 formulated as [Cu(H2L13)2]n (H3L13 = 2-ethyl-1H-imidazole-4,5-dicarboxylate), prepared by the hydrothermal reaction of CuCl2 with H3L13. They were obtained from the same reaction, but at different temperatures, i.e., 160 °C and 120 °C, respectively.
Single crystal X-ray structural analysis revealed that both frameworks have two-dimensional square grid-like layers with a uniform (4,4) net topology. They crystallize in the monoclinic P21/n and P21/c space groups, respectively, and each repeated unit consists of one Cu2+ ion and one μ2-H2L13 ligand. For both frameworks, the metal centre is hexa-coordinated and surrounded by two N-atoms and four O-atoms from four H2L13 ligands. The neighbouring Cu⋯Cu distances are 8.639 Å and 6.138 Å, respectively.
Kitagawa et al. reported30 three genuine supramolecular isomers (30–32) with the formula [Cu2(OAc)2(L14)]n (L14 = benzoquinone, OAc = acetate) which were synthesized by the reaction of copper(II) acetate with hydroquinone in ethanol at different temperatures. Framework 30 (α phase) was prepared at 30 °C, but 31 (β phase) and 32 (γ phase) were isolated at 40 °C and 80 °C, respectively.
Single crystal X-ray structural analysis revealed that 30 and 31 have 2D structures, whereas 32 is one-dimensional. They have crystallized in different space groups and the {Cu2(OAc)2(L14)} dimeric unit acts as a SBU, being interconnected through oxygen atoms from L14 or acetate ligands giving rise to 1D or 2D structures.
In compound 30, the dinuclear Cu(I) units are interconnected via oxygen atoms from L14 ligands, resulting in the formation of a 2D sheet. The L14 and AcO− ligands are coordinated to the Cu(I) centres in a μ4 and μ2 fashion, respectively. For compound 31, the axial positions of the dicopper core are coordinated by O atoms from L14 and AcO− ligands and construct a 2D infinite structure. In this compound, the benzoquinone ligand functions as a μ3-bridging ligand, and two AcO− ligands act as μ2 and μ3 bridging ligands. For 32, the axial positions of the dicopper unit are coordinated by two AcO− ligands forming a 1D chain. In this compound, the acetate ligand acts as a μ3-bridge.
Another example of temperature-dependent structural isomerism was reported by Ghosh et al.31 They have synthesized three anionic MOFs (33–35), with the general formula [Me2NH2]n[Zn1.5(L15)2]n, by the solvothermal reaction of the Zn(II) salt and H2L15 (H2L15 = 2,5-furandicarboxylic acid) in DMF at different temperatures, i.e., 90 °C, 120 °C or 160 °C, respectively.
The crystal structure analysis of these frameworks revealed that the asymmetric unit of these frameworks contains two types of Zn(II) centres (one with full and the other with half occupancy), two L15 ligands and one dimethyl ammonium (DMA) cation. The metal centres are interconnected by bridging carboxylate units, forming a linear trinuclear Zn(II) cluster, with different coordination environments, which behaves as a SBU. In 33 and 34, one trinuclear unit acts as a SBU, but in 35, two different trinuclear units act as such.
For framework 33, one Zn(II) centre has a distorted octahedral geometry but the other one has a perfect octahedral coordination. Interestingly, with increasing reaction temperature for compounds 33 and 34, from 90 °C to 120 °C, the octahedrally distorted Zn(II) centre in 33 transformed to tetrahedral in one trinuclear unit and distorted trigonal bipyramidal in another trinuclear unit for 34. But upon further rise in temperature to 160 °C, the perfect octahedral Zn(II) centre in 33 converted to a tetrahedral one for framework 35.
In framework 33, each trinuclear cluster is connected to four such clusters by a pair of L15 ligands on each side and extended into a 2D net in the ab plane. But in the case of 34, two types of trimeric units are connected by eight L15 ligands which are further interconnected by other trimeric units, forming a 3D structure. For 35, the trimeric units are connected by four L15 ligands in the bc plane to form a 2D sheet. Adjacent 2D sheets are connected at the trimeric units from both sides by a pair of L15 ligands, leading to a 3D framework.
2.3 pH-Dependent structural isomerism
Reaction pH can also play an important role in the formation of different structural isomers, as reported by Lei et al.32 They have synthesized the structural isomers [Co(L16)(4,4′-bibp)]n (36) and [Co(L16)(4,4′-bibp)]n·2nH2O (37) by the hydrothermal reaction of cobalt(II) nitrate hexahydrate, H2L16 [3,5-bis(3-carboxyphenyl)pyridine] and 4,4′-bibp [4,4′-bis(imidazol-1-yl)biphenyl] at different pHs (Fig. 9). Compound 36 does not have any solvent of crystallization, while 37 has two molecules of water. | ||
| Fig. 9 Synthesis and crystal structures of compounds 36 and 37. Reprinted (adapted) with permission from ref. 32. Copyright 2013 American Chemical Society. | ||
The structural analysis revealed that 36 and 37 have two- and three-dimensional structures with (3,5)-connected (42·67·8)(42·6) 3,5-L2 and (4,6)-connected (44·610·8)(44·62) fsc topologies, respectively. The asymmetric unit of 36 contains one Co(II) ion, one L16 ligand and one 4,4′-bibp ligand, and the metal centre has a distorted heptahedral geometry. The Co(II) ions are bridged by completely deprotonated L16 ligands, forming a 1D ladder-like structure which is further interconnected by 4,4′-bibp to construct a 2D network. Compound 37 has a similar asymmetric unit, but with two non-coordinated water molecules, and its Co(II) ion is hexa-coordinated by two N-atoms from two 4,4′-bibp ligands and one N- and three O-atoms from four L16 ligands. The L16 ligands coordinate to the Co(II) ions in such a way that left- and right-handed helical chains are constructed along the crystallographic a-axis, being further connected by 4,4′-bibp to form a 3D structure. These two compounds provide an example of structural isomerism induced by the pH of the reaction medium.
2.4 Crystallization method-dependent structural isomerism
In some cases, the crystallization conditions become a key factor in forming different supramolecular isomers. For example, Tao et al. reported33 two 3D supramolecular isomers with the molecular formula [Mn(L17)(H2O)2]n·nH2O (38 and 39), derived from the one-dimensional CP [Mn(L17)(H2O)(CH3OH)]n (H2L17 = L-malic acid) by using different methods of crystallization.The reaction of MnCl2·4H2O with H2L17 in a mixture of CH3OH–H2O and triethylamine led to the formation of the 1D MOF [Mn(L17)(H2O)(CH3OH)]n. When this compound was kept in solution, it dissolved in one day and converted to crystals of 38 after two days. However, when [Mn(L17)(H2O)(CH3OH)]n was dissolved slowly in a sealed container, it converted to pink prism-like crystals of 39 in two months (Fig. 10). The crystallization of 38 and 39 was thermodynamically controlled, but the crystallization of [Mn(L17)(H2O)(CH3OH)]n was kinetically controlled.
 | ||
| Fig. 10 3D frameworks of compounds 37 and 38. Reprinted (adapted) with permission from ref. 33. Copyright 2008 Royal Society of Chemistry. | ||
The crystal structure analysis showed that 37 and 38 are structural supramolecular isomers with an identical chemical composition but they have crystallized in different space groups, P212121 and P43, respectively. In both of them, the Mn(II) centres have a distorted octahedral geometry and are coordinated by three carboxylate oxygen atoms, one hydroxyl O-atom and two water molecules in cis (for 37) and trans (for 38) fashions. This slight difference results in quite different architectures. The 3D polymeric structure of 37 has 1D channels running along the c-axis, whereas for 38 the 3D framework shows rectangular channels with S4 symmetry. These MOFs constitute an example of structural isomerism which is controlled by the method of crystallization.
Another example of crystallization method-dependent structural supramolecular isomers was reported by Sun and co-workers.34 They have synthesized two 1D nanotubular supramolecular isomers, [Zn(L18)(bipy)]n·3nH2O (39 and 40), by self-assembly of 5-amino-2,4,6-triiodoisophthalic acid (H2L18), 4,4′-bipyridine (bipy) and Zn(II) ions at room temperature. Light yellow prismatic crystals of 39 were formed after one week from the reaction mixture. However, keeping the mixture in a sealed container for 4 weeks resulted in the formation of 40. The structural analysis revealed that both frameworks crystallize in the same space group and have the same asymmetric unit. The most significant structural change is that for 39 the 4,4′-bipyridine ligands connect Zn(II) ions to form 0D Zn–bipyridine squares which are further interconnected by L18 ligands forming 1D nanotubes. However, for 40, the connection of Zn(II) ions to 4,4′-bipyridine generates a 1D Zn–bipy helical chain which is further bridged by the L18 ligand to form two 1D nanotubular supramolecular isomers (Fig. 11).
 | ||
| Fig. 11 0D square (a) and 1D helical (b) structures in compounds 39 and 40 constructed from Zn(II) and 4,4′-bipyridine, respectively. 1D nanotubes along the c-axis: (c) 39; (d) 40. Nanotubular channels of 39 (e) and 40 (f) are shown by the yellow tubes. Reprinted (adapted) with permission from ref. 34. Copyright 2009 American Chemical Society. | ||
2.5 Template-induced structural isomerism
Template-induced structural isomerism is not very well known (only scant examples have been reported) and the exact role of the template is generally uncertain. The template effect can be associated with the size compatibility between the framework pores and the template. Banerjee et al. have reported three Mn(II)-based MOF isomers (41–43) which were synthesized by using different organic molecules as templates.35 Framework 41 was obtained by the reaction of 5-triazole isophthalic acid and Mn(NO3)2·xH2O at 85 °C for 72 h. The two other isomers, 42 and 43, were obtained by a similar reaction, but in the presence of pyrazine and 4,4′-bipyridine, respectively (Fig. 12). It is noteworthy to mention that in the MOFs the template (pyrazine or 4,4′-bipyridine) is neither coordinated to metal ions nor present inside the pores of the frameworks. | ||
| Fig. 12 Synthesis and space-filling models of frameworks 41–43. Reprinted (adapted) with permission from ref. 35. Copyright 2011 Royal Society of Chemistry. | ||
The crystal structure analysis revealed that all these MOFs have 3D structures with an octahedral (for 41–43) or a trigonal bipyramidal (for 41, one Mn ion) coordination geometry. The isomer 41 is nonporous, while for 42 and 43 the pore size increases to 2.56 Å and 7.26 Å, respectively, which are comparable to the length of pyrazine (2.81 Å) and 4,4′-bipyridine (7.11 Å). Framework 43 also has a higher solvent-accessible void (52.5%) than 42 (51.6%). Structural conversions from non-porous to porous due to the template effect during the synthesis have been observed.
3. Conformational isomerism
Conformational isomers of MOFs contain the same building block units and ligands but they differ either by the conformation of flexible ligands or by the orientation of such units.36 Many external factors, such as the temperature, solvent, concentration and template, may result in conformational isomerism of coordination networks.10,11 In the following section, we present selected examples of MOFs having conformational isomers.3.1 Solvent-dependent conformational isomerism
Solvent can play an important role in the ligand conformation and thus in solvent-induced conformational isomerism, as illustrated below.Paine et al. have reported37 three isomeric silver(I) 1D MOFs (44–46) having the general formula [Ag(L19)]n(ClO4)n. The ligand L19 is a bis(bidentate) N,N-donor Schiff-base which was prepared by the condensation of α,α′-diamino-p-xylene and pyridine-2-carboxaldehyde, and the MOFs were synthesized by its reaction with anhydrous silver perchlorate. Isomers 44 and 45 were obtained upon crystallization of the bulk product from acetonitrile. Initially, prism-shaped crystals (44) were isolated followed by the isolation of rhombic-shaped crystals (45) from the filtrate. But compound 46 crystallised during the synthesis from a solvent mixture of dichloromethane and methanol. Therefore, in these cases, the solvent plays a vital role in the formation of the isomers.
Single-crystal X-ray structure analysis revealed that all these MOFs have 1D structures where the silver ions display a pseudotetrahedral N4 coordination geometry (Fig. 13). They crystallized in different space groups, i.e., monoclinic Pc, Cc and C2/c, respectively. Compound 44 comprises two isomeric and parallel 1D polymeric chains of L19 and Ag(I) ions. The asymmetric unit of 45 contains two polymeric chains, perchlorate anions, acetonitrile and water molecules. But the asymmetric unit of compound 46 contains two bis(bidentate) ligands and two silver(I) ions without any lattice solvent. Isomer 46 has a 1D chain like 45, which is further extended along the diagonal axis of the ac plane.
 | ||
| Fig. 13 Synthesis and 1D structures of compounds 44–46. Reprinted (adapted) with permission from ref. 37. Copyright 2011 Royal Society of Chemistry. | ||
The major difference in these three isomers arises from the different conformations of the flexible ligand L19. In these isomers, the ligand L19 adopts the conformations gauche and anti and both conformers exist in equilibrium due to the low energy barrier for the conformational change. For compound 44, both gauche and anti conformations of the ligand are found, but for 45 and 46, only the anti-conformation is observed. Thus, the isomerism arises from the different conformations of L19 and the MOFs are conformational supramolecular isomers.
Another example of solvent-induced conformational isomerism was reported by Vittal and co-workers,38a who have synthesized the conformational isomers [Zn2(L20)(L21)2]n·5nH2O (47) and [Zn2(L20)(L21)2]n·2nDMF·nH2O (48) by using different solvent systems. The solvothermal reaction of zinc(II) nitrate, 1,4-bis[2-(4-pyridyl)ethenyl]benzene (L20) and 4,4′-oxybisbenzoic acid (H2L21) in DMSO in the presence of dimethylacetamide (DMA) produces a threefold interpenetrated 3D porous MOF (47) having the pcu-type topology (Fig. 14A). However, the use of N,N-dimethylformamide (DMF) instead of DMA generates another isomer, 48. Single-crystal X-ray diffraction analysis revealed that 47 and 48 crystallised in the monoclinic C2/c and P21/c space groups, respectively. Both of them have the same asymmetric unit which contains two Zn(II) atoms, two L21 ligands and one L20 ligand. But they have different solvents of crystallization (five water molecules for 47 and 2 DMF molecules and one water molecule for 48). For both frameworks, the Zn(II) ion has a distorted square-pyramidal geometry, coordinated by four oxygen atoms from three L21 ligands at the equatorial positions and one nitrogen atom of the L20 ligand at the axial position. A paddle-wheel-type Zn(II) dinuclear cluster acts as a SBU for both frameworks, being interconnected via the L20 ligand which acts as a pillar and produces a pillared-layer structure.
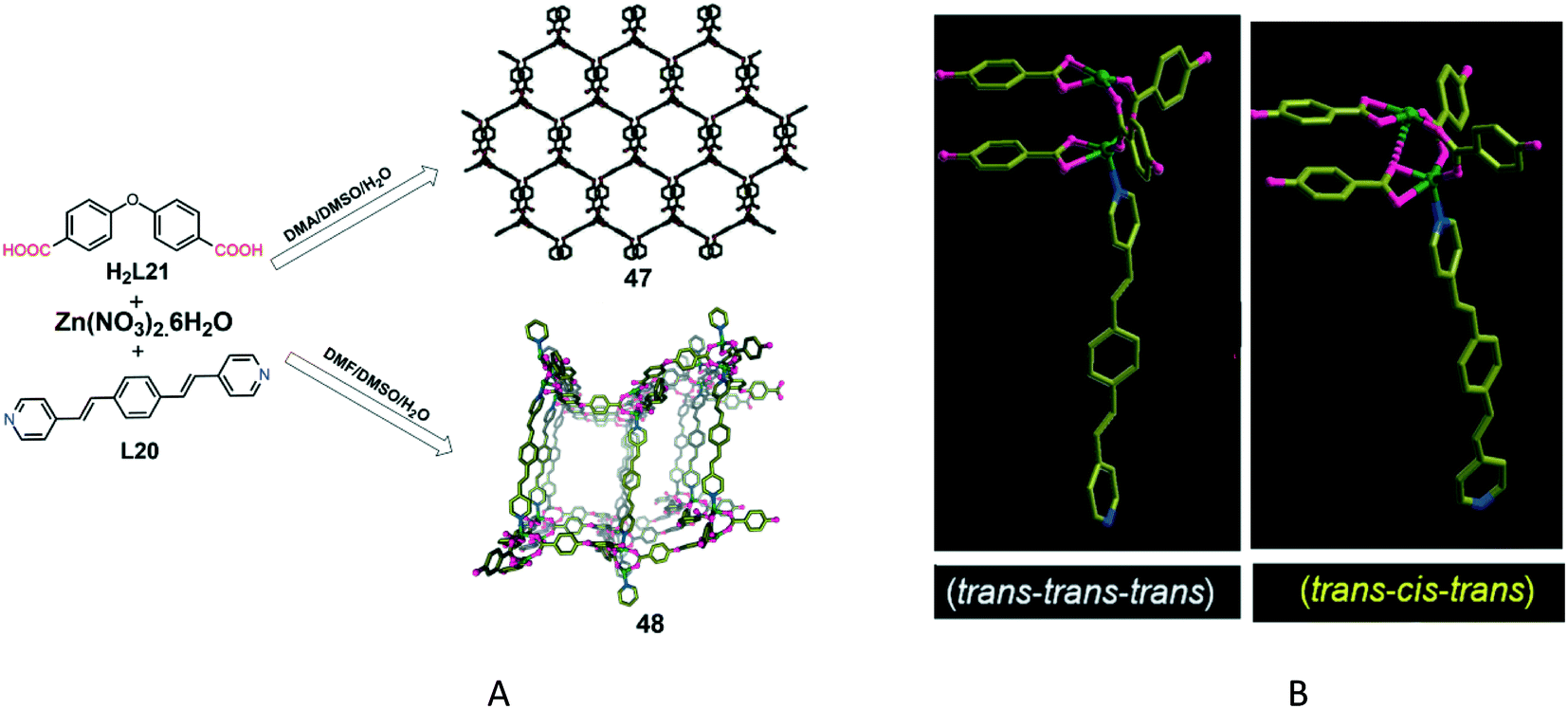 | ||
| Fig. 14 A) Synthesis and crystal structures of frameworks 47 and 48. B) Different orientations of 1,4-bis[2-(4-pyridyl)ethenyl]benzene in compounds 47 (trans–trans–trans conformation) and 48 (trans–cis–trans conformation). Reproduced with permission from ref. 38a. Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
The most significant difference between these two MOFs concerns the conformation of the L20 ligand. In 47, this ligand shows a trans–trans–trans conformation, whereas in 48, it has a trans–cis–trans orientation (Fig. 14B). The different conformations of the L20 ligand account for the overall structural difference between the two isomeric frameworks.
Another example of solvent-dependent conformational isomerism was indicated by Ghosh et al.38b They have reported two MOFs (49 and 50) synthesized by the reaction of Cd(II) ions and 1,4-bis(4-pyridyl)-2,3-diaza-1,3-butadiene (L22) in different solvents. The conformational isomerism arises mainly due to conformation flexibility of the ligand (L22). The reactions of Cd(ClO4)2 with L22 in a mixture of CH2Cl2/toluene/MeOH gave the framework [Cd(L22)2(ClO4)2]n·n(toluene)·n(MeOH) (49), whereas by performing the reaction by using mesitylene instead of toluene, the other isomer [Cd2(L22)4(ClO4)4]n·3n(mesitylene) (50) was obtained.
The structural analysis of these frameworks revealed that both of them have 2D structures. In 49, the asymmetric unit contains one Cd(II) ion, two L22 molecules, two ClO4− ligands, and one each of non-coordinated toluene and methanol molecules. However, for 50, the asymmetric unit contains two Cd(II) ions, four L22 and four perchlorate ligands, apart from three non-coordinated mesitylene guest molecules. The perchlorate anions coordinate to the metal centre in a monodentate (for 49 and 50) or bidentate (for 49) fashion. For compounds 49 and 50, the Cd(II) centre is hepta- and hexa-coordinated, respectively. In 49, one ligand (L22) is in the trans form and the other one is in the cis form, but for 50, all L22 ligands are in the trans conformation. For both frameworks, the L22 ligand coordinates to the metal via the terminal nitrogens, thereby extending the frameworks to form 2D nets. For 49, the nets are not planar due to the cis geometry of one of the ligands, but in 50, the trans character of the ligands facilitates the formation of 2D planar sheets.
Thus, MOFs 49 and 50 constitute a good example of conformational supramolecular isomers, which arise from the combined effects of ligand conformations and solvents used for their synthesis.
3.2 Temperature-dependent conformational isomerism
Reaction temperature can play an important role in the assembly process of the metal and ligand and can constitute an important factor for the construction of conformational isomers.An example of temperature-dependent conformational isomerism was reported by Zhou et al.39 They have synthesized two conformational isomers, 50 and 51, with a tetra-carboxylate ligand [N,N,N′,N′-tetrakis(4-carboxyphenyl)-1,4-phenylenediamine (L23)], formulated as [Cu2(L23)(H2O)2]n·2nDMSO·6nH2O. The hydrothermal reaction of H4L23 and Cu(NO3)2·2.5H2O in DMSO at 120 °C gave 50, but when the reaction was carried out at 115 °C, it produced the isomer 51 (Fig. 15c and d).
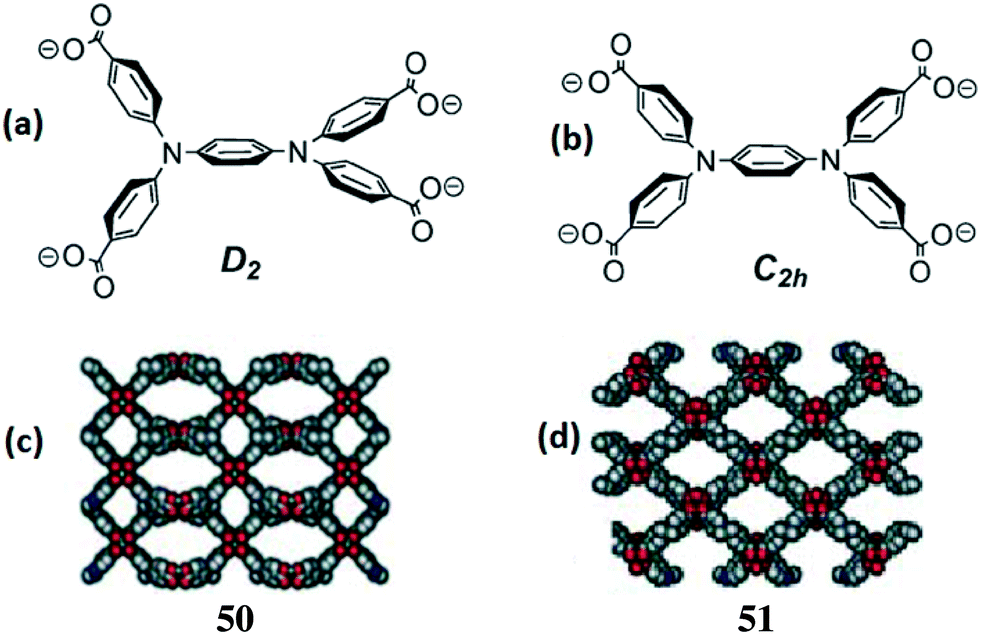 | ||
| Fig. 15 Different orientations [D2 (a) and C2h (b)] of ligand L23 and the crystal structures of 50 (c) and 51 (d). Reprinted (adapted) with permission from ref. 39. Copyright 2005 Royal Society of Chemistry. | ||
X-ray diffraction analysis revealed that 50 and 51 crystallized in orthorhombic Cccm and Imma space groups, respectively. In both MOFs, a paddle-wheel-type Cu(II) dinuclear cluster acts as a SBU and each copper ion adopts a square-pyramidal coordination geometry. The equatorial positions of the dinuclear copper unit are connected by four carboxylate groups from four different L23 ligands and the axial positions are occupied by water molecules. Each L23 ligand is coordinated to four dinuclear copper SBUs and generates porous 3D frameworks having PtS (for 50) and NbO (for 51)-type nets. In these frameworks, the flexible tetracarboxylate ligand adopts two different interconvertible conformations, i.e., a tetrahedral D2 conformation in the case of 50 and a square-planar C2h conformation for 51 (Fig. 15a and b). Due to the small energy barrier between the two isomeric forms of the ligand, they were not separated at room temperature, but they were stabilized and isolated in their pure form in the coordination networks. A N2 adsorption study of these MOFs shows that they possess type-I isotherms with Langmuir surface areas of 626.72 (for 50) and 504.22 m2 g−1 (for 51).
Another set of temperature-dependent conformational isomers concerns the 3D frameworks of copper [Cu6(L24)3(H2O)6]n·3nDMA·6nH2O (52) and [Cu2(L24)(H2O)2]n·3nDMSO (53), obtained via the solvothermal reaction of Cu(NO3)2·2.5H2O and 5,5′-methylene-di-isophthalic acid (H4L24) in dimethylacetamide (DMA) at 85 °C and DMSO at 120 °C, respectively (Fig. 16).40 The primary factor for the structural difference between the two structures was the temperature of the solvothermal reaction. Although the solvent can play an important role in the formation of different frameworks, in this case it seems to have a secondary role towards the conformational isomerism. Single-crystal X-ray analysis revealed that 52 crystallized in the tetragonal P4/mmm space group and 53 crystallized in the P63/mmc space group. The two frameworks are composed of paddlewheel-type dinuclear copper(II) units and 5,5′-methylene-diisophthalate (L24) as the building block units with water molecules coordinated in the axial position of the paddlewheels. Moreover, the flexible nature of the ligand leads to differences in topologies of the extended structure.
 | ||
| Fig. 16 Synthesis and open metal site alignment of the MOF polymorphs 52 and 53. Reproduced with permission from ref. 40. Copyright 2008 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
The conformational polymorphism arises from the different conformations of the L24 ligand, one with the Cs symmetry, in which the two phenyl rings of L24 are perpendicular to each other, and another one with the C2v symmetry. In 52, two L24 ligands have Cs symmetry and one has C2v, but in 53, all L24 ligands only have C2v symmetry.
A hydrogen adsorption study of 52 shows a remarkably high hydrogen uptake of 3.05 wt% (23.2 g L−1) at 77 K and 1 atm, whereas 53 shows a 2.40 wt% (20.4 g L−1) hydrogen uptake under the same conditions. A similar phenomenon was observed by performing a nitrogen adsorption study. The Langmuir surface area of 52 is 2425 m2 g−1, whereas that of 53 is 1962 m2 g−1. The higher gas adsorption in framework 52 is mainly due to its close packing type of structure which contains cuboctahedral cages and the arrangement of open metal sites within these cages.
Another example of conformational framework isomers was reported by Férey et al. (Fig. 17).41 They have synthesized a 3D Cr(III) MOF (54) by the hydrothermal reaction of Cr(NO3)3 with terephthalic acid in water in the presence of hydrofluoric acid. This MOF shows a high breathing effect (reversible change of structure and shape of its pores) during the hydration and dehydration process. When it was heated at 300 °C, a calcined form (54ht) was obtained which upon cooling in air formed another isomeric form (54lt). Both 54 and 54ht crystallized in orthorhombic space groups having very similar unit cell parameters, but 54lt crystallized in the monoclinic C2/c space group. These frameworks exhibit a similar type of 3D structure constructed from chromium(III) octahedra and terephthalate ions having 1D porous channels. MOFs 54 and 54ht have similar types of square channels with approximate dimensions of 17 × 13 Å2. However, in the case of 54lt, the channels' shape is a rhombus with an approximate size of 19 × 8 Å2. Thus, these compounds are examples of conformational isomers which arise mainly from different coordination parameters of the metal site and not from different conformations of the ligand.
 | ||
| Fig. 17 Crystal structures of 54 (a), 54ht (b) and 54lt (c). Reprinted with permission from ref. 41. Copyright 2002 American Chemical Society. | ||
Xing et al. reported four Cd-MOFs, formulated as (Me2NH2)4n[Cd2(L25)2]n·2nDMF (55), (Et2NH2)2n[Cd(L25)]n·nH2O (56), (Et2NH2)3n[Cd2(L25)2]n·2nDEF·4nH2O (57; DEF = diethylformamide) and (Et2NH2)3n[Cd2(L25)2]n·2nEtNH2·2nH2O (58) [H4L25 = 5-[bis(3-carboxybenzyl)amino]isophthalic acid], which are conformational supramolecular isomers (Fig. 18).42 They were synthesized by the solvothermal reaction of Cd(NO3)2 and 5-[bis(3-carboxybenzyl)amino]isophthalic acid in diethylformamide (DEF)/H2O at different reaction temperatures. Framework 56 was prepared at 110 °C, while 57 and 58 were obtained at 140 °C and 150 °C, respectively. However, 55 was synthesized by using a different solvent system (DMF/H2O) at 110 °C. Single-crystal X-ray diffraction analysis revealed that these MOFs crystallized in the same monoclinic P21/c space group and have a similar type of 2D double-layered structure with (4,4)-network topology. In MOFs 55–57, the Cd(II) centres are seven-coordinated by oxygen atoms from four ligands and produce a distorted pentagonal bipyramidal geometry. However, for 58, the metal centre has an octahedral coordination geometry with two monodentate and two chelating carboxylate groups. These MOFs have similar compositions but the conformations of the ligands are different.
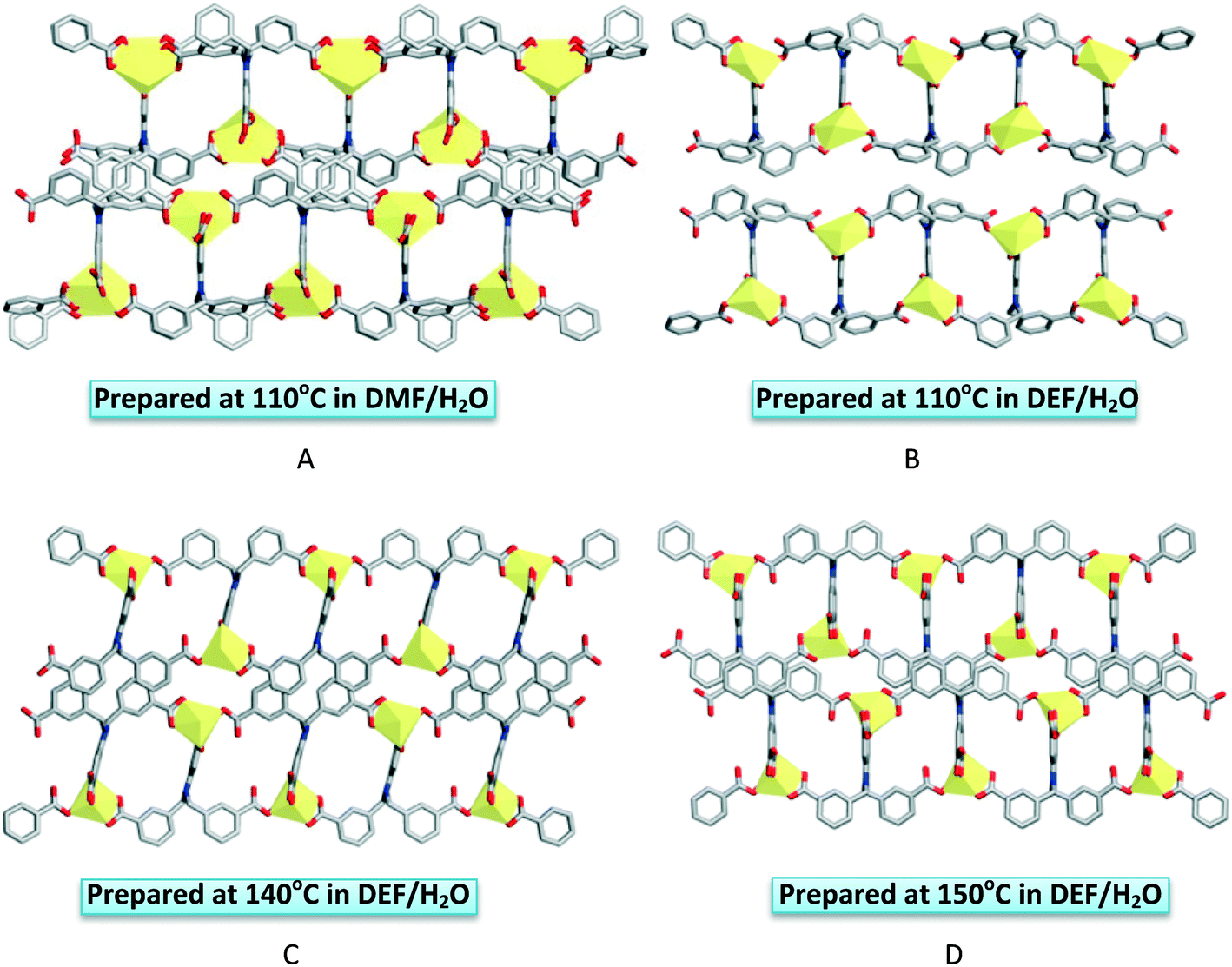 | ||
| Fig. 18 Presentation of the layered structures of 55 (A), 56 (B), 57 (C) and 58 (D). Reprinted (adapted) with permission from ref. 42. Copyright 2013 American Chemical Society. | ||
3.3 Photo-induced conformational isomerism
An example of conformational isomerism triggered by UV light was recently reported by Zhao and co-workers.43a They have synthesized a self-catenated three-dimensional Zn-MOF formulated as [Zn2(L26)(L21)2]n·nDMF (59) by the solvothermal reaction of Zn(NO3)2·6H2O, 3,3′-bis[2-(4-pyridyl)ethenyl]azobenzene (L26) and 4,4′-oxybisbenzoic acid (H2L21) (Fig. 19A). Single crystal X-ray analysis revealed that the asymmetric unit of 59 contains one Zn2+ ion, half L26, one L21 ligand and half DMF molecule. Each Zn(II) centre adopts a tetragonal pyramidal coordination geometry, being coordinated by four oxygen atoms from four L21 ligands and one N-atom from ligated L26. In this framework, the dinuclear {Zn2(COO)4} unit acts as a SBU which is connected to four L21 ligands forming a 2D layer structure with rhombic grids. These 2D layers are interconnected by L26 ligands and construct a pillared-layer type of porous 3D framework. Topological analysis revealed that this framework is a six-connected uninodal net with a Schläfli symbol of 44·610·8. | ||
| Fig. 19 A) A perspective view of the 3D structure of 59. B) Light-triggered single crystal-to-single crystal (SCSC) transformation through pedal motion in 3,3′-bis[2-(4-pyridyl)ethenyl]azobenzene. Reprinted (adapted) with permission from ref. 43a. Copyright 2016 Nature Publishing. | ||
Upon exposure to UV light, 59 gives rise to its conformational isomer 59_UV. In the latter, a pedal motion along the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– and –NN– moieties of L26 ligands is observed (Fig. 19B). However, the overall connectivity of this framework remains as in 59. Only slight changes in bond distances and angles between the two frameworks occur, mainly on account of the conformational changes of the L26 ligand upon exposure to UV light.
C– and –NN– moieties of L26 ligands is observed (Fig. 19B). However, the overall connectivity of this framework remains as in 59. Only slight changes in bond distances and angles between the two frameworks occur, mainly on account of the conformational changes of the L26 ligand upon exposure to UV light.
A gas adsorption study showed that the activated framework 59 adsorbs a higher amount of CO2 than N2 and exhibits a normal type-I isotherm having a BET surface area of 299 m2 g−1. It can adsorb 1.69 mmol of CO2 per g at 298 K and 120 kPa, but under similar conditions, 59_UV can adsorb up to 1.43 mmol of CO2 per g. The authors have also studied the detection of nitroaromatic explosives in water medium.
Another interesting example of photo-induced conformational isomerism was reported by Zhou and co-workers.43b They have synthesized a 3D Zn(II) MOF (60) (the structure of which was not established by single crystal X-ray diffraction analysis) by the hydrothermal reaction of 2-(phenyldiazenyl)terephthalic acid (H2L27) and zinc nitrate in diethylformamide (DEF) at 85 °C. This acid was chosen as a linker due to its isomerization capability under UV light (from trans to cis) and thermal conditions (from cis to trans) (Scheme 2A).
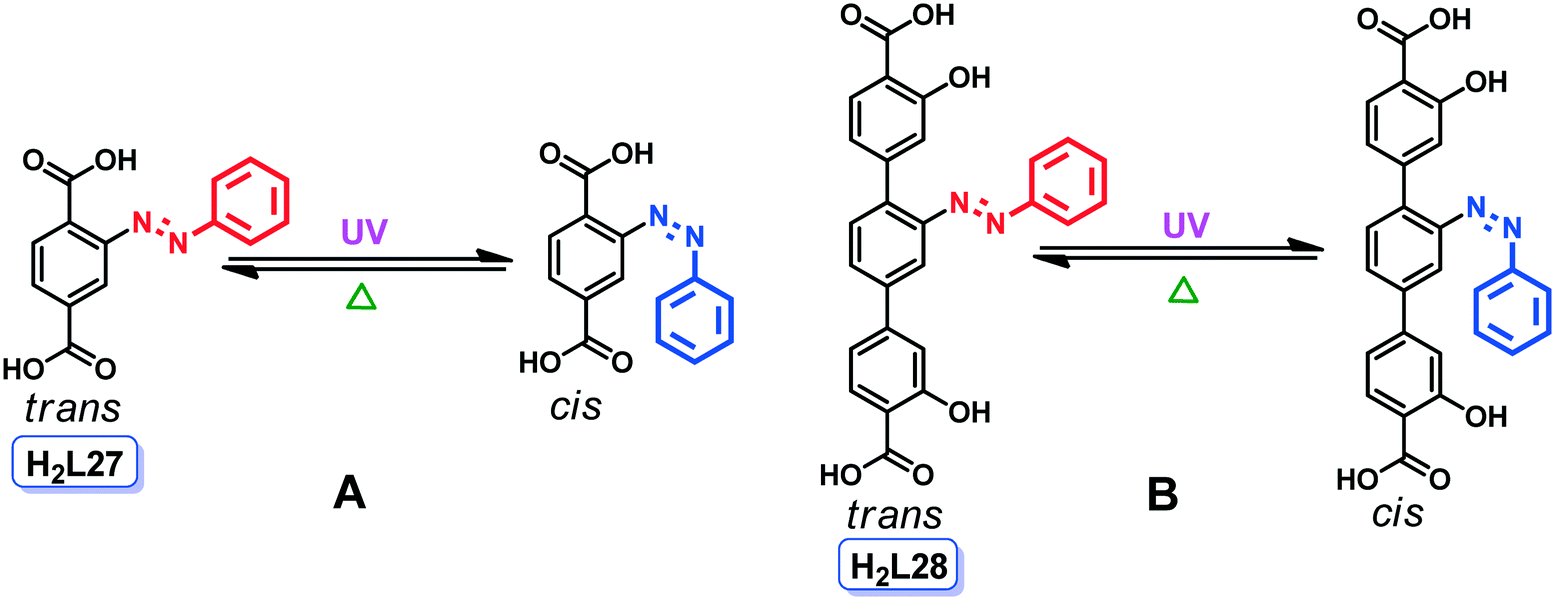 | ||
| Scheme 2 Isomerization of pro-ligands H2L27 (A) and H2L28 (B) in the presence of UV and heat. | ||
In 60, the azobenzene group of L27 is mostly present in the trans orientation, but upon exposure to UV light, the conformation of the ligand changes to cis, producing the new conformational isomer 60_UV. In these frameworks, the isomerism mainly arises from the conformational change (trans-to-cis) in the azobenzene unit (Scheme 2A). This small structural transformation in the ligand unit makes a significant impact on the gas adsorption behaviour of these frameworks. In 60, the CO2 uptake at room temperature after activation is 22.9 cm3 g−1 at 1 bar, but after 1 h of UV irradiation, the uptake decreases to 16.8 cm3 g−1. The uptake further decreases to 10.5 cm3 g−1 (upon measuring the sample for the 2nd time) which corresponds to an overall decrease of 54% in CO2 uptake compared to 60. The slow change in gas adsorption isotherms upon UV irradiation may be due to the slow isomerization of the ligand (L27) in the framework. The azobenzene groups in 60 are predominantly in the trans conformation, and after UV irradiation for 1 h, only a few of them become cis. This conformational change triggers the whole process and more trans isomers convert to cis, leading to a significant decrease in gas uptake.
Similar types of conformational isomers were also reported by Yaghi and co-workers.43c They synthesized an azobenzene-functionalized isoreticular Mg-MOF (61) (the structure of which was not established by single crystal X-ray diffraction analysis) by combining Mg(NO3)2·6H2O and an azobenzene-functionalized linker (L28) (for the formula, see Scheme 2B) in a mixture of DMF–EtOH–H2O at 120 °C. In this framework, the Mg(II) ions are hexa-coordinated by carboxylate linkers forming a 3D network with 1D hexagonal pores. The framework 61 has an etb topology with a BET surface area of 2410 m2 g−1.
In 61, the linker L28 is ornamented with a photo-switchable azobenzene unit, which can be switched from trans to cis or cis to trans using UV or visible irradiation (Scheme 2B), respectively. These conformational changes in the azobenzene unit lead to the formation of two different conformational isomers, 61_cis and 61_trans, where all of the azobenzene units are in the cis and trans conformation, respectively. 61_cis switches to 61_trans upon excitation at 408 nm and the pore size decreases from 10.3 Å to 8.3 Å. This transformation was monitored by studying the storage and release capabilities of propidium iodide dye molecules in the different isomers. Due to the higher pore diameter of 61_cis, it is able to store the propidium iodide dye molecules in its 1D channel. But upon irradiation at 408 nm, the rapid conversion to 61_trans prompts the release of the dye from the pores.
3.4. Additive-induced conformational isomerism
In some cases, additive or template molecules can play a critical role in the formation of different supramolecular isomers. We now present a few examples of conformational isomers where different additives or templates significantly affect the conformation of the ligands as well as the overall topology of MOFs.Du and co-workers reported an example of additive-induced conformational isomerism.44 They synthesized the isomers [Ag(L29)]2(NO3)2 (62) and [Ag(L29)]n(NO3)n (63) by the hydrothermal reaction of 3,5-bis-(2-pyridyl)-4-amino-1,2,4-triazole (L29) and AgNO3 in the presence of different organic acids as additive agents. For their synthesis, adipic acid and D,L-mandelic acid (length of ca. 9.8 Å and 5.5 Å, respectively, calculated by using the Mercury 3.9 crystallographic software) were used as additives (Fig. 20).
 | ||
| Fig. 20 The crystal structures of supramolecular isomers 62 (the dinuclear motif) and 63 (the 1D helical chain). Reprinted (adapted) with permission from ref. 44. Copyright 2013 American Chemical Society. | ||
Single crystal structural analysis revealed that compounds 62 and 63 have a dinuclear and a 1D helical-type structure, respectively. In both compounds, the coordination geometry of the Ag ion and the binding mode of the L29 ligand are similar. The asymmetric unit of both compounds contains one silver ion, one L29 ligand and one nitrate anion remains as a counter ion. Each metal centre adopts a distorted square planar geometry with coordination sites being occupied by four N-atoms from two L29 ligands. For these compounds, the isomerism arises predominantly from different orientations of the L29 ligands. In 62, the dihedral angle between two 2-pyridyl rings in L29 is 3.6°, but in the case of 63, a higher planarity in this ligand is observed, the dihedral angle between the two 2-pyridyl groups being 1.3°. These isomers are also interconvertible to each other.
Solvent molecules can display an important role in the construction of supramolecular isomers. In some cases, the solvent can act as a template for the synthesis of conformational isomers. An example has been reported by Hou's group.45 The conformational isomers [Co2(L30)(CH3CN)(H2O)3]n·nCH3OH·nH2O (64) and [Co2(L30)(CH3CN)(H2O)3]n (65) [H4L30 = 5,5′-(pentane-1,2-diyl)-bis(oxy)diisophthalic acid] were prepared under the same solvothermal conditions, but with different concentrations of a cyclic ether as a template agent.
MOF 64 was synthesized by the reaction of Co(NO3)2·6H2O and Na4L30 in a mixture of acetonitrile–methanol–water at 160 °C for 3 days. However, in the presence of a low concentration of 1,4-dioxane as a template, compound 65 was obtained (Fig. 21). Single crystal X-ray diffraction analysis revealed that 64 and 65 crystallized in the monoclinic P21/c and triclinic P space groups, respectively. The asymmetric unit of both frameworks is the same and consists of two Co(II) ions, one L30 ligand, three water and one acetonitrile molecules. In both cases, the dinuclear {Co2(COO)2} unit acts as a SBU, being additionally connected by L30 ligands to construct two-dimensional (for 65) and three-dimensional (for 64) structures. In both compounds, the structural formula and the connectivity are almost identical, except for the orientation of the L30 ligand. The dihedral angle between the two aromatic rings of the L30 ligand is 20.0° and 0.3° for 64 and 65, respectively, thus the ligand being more planar in the latter case. The addition of a small amount of 1,4-dioxane led to a change of the 3D structure of 64 to a 2D layer structure of 65, with the channel size increasing from ca. 6 × 6 Å2 to ca. 15 × 8 Å2. The conformational change of the L30 ligand in 65 compared to 64 may be due to the interactions, during the synthesis, between the electronegative O-atoms of the cyclic ether (1,4-dioxane template) and the electron-deficient aromatic unit (electron-deficient π-system) of the L30 ligand.
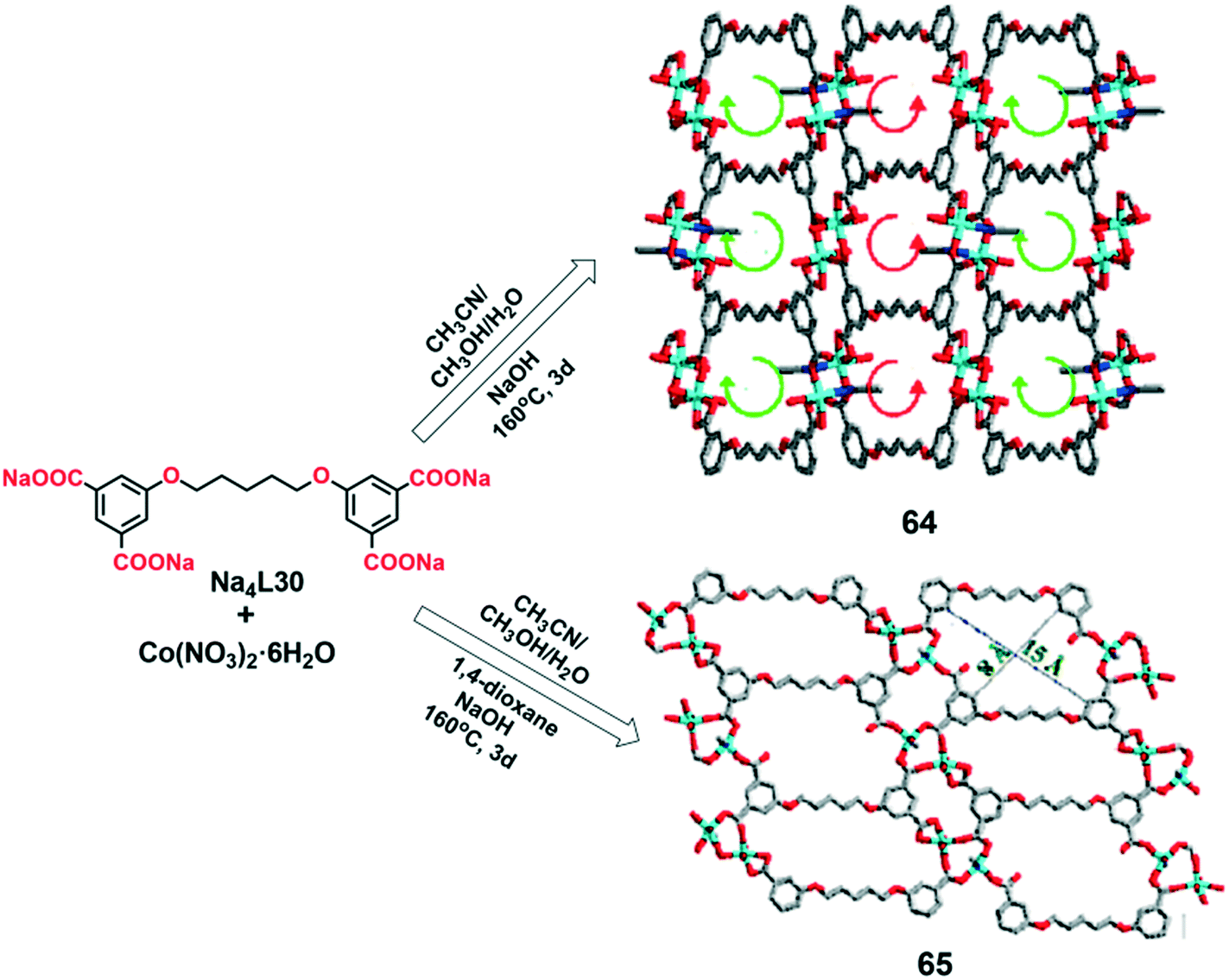 | ||
| Fig. 21 Synthesis and crystal structures of MOFs 64 and 65. Reprinted (adapted) with permission from ref. 45. Copyright 2015 American Chemical Society. | ||
These MOFs catalyze cascade cycloaddition reactions of aromatic nitriles with diamines, where the catalytic activity of 65 was much higher than that of 64 on account of its unique structural features, accessible catalytic sites and suitable channel size and shape.
3.5 Reaction condition-dependent conformational isomerism
Conformational isomerism of MOFs is typically caused by variations of the metal coordination geometry or of the coordination and conformation of ligands. Many factors, e.g., the temperature, solvent, template and reaction conditions, can affect the self-assembly of supramolecular isomers.10,11,16 We have already discussed the effect of temperature,39–42 solvent,37,38 UV-light43 and additives44,45 on the conformational supramolecular isomerism of various MOFs and now we are going to illustrate how other different reaction conditions, namely the reagents' molar ratio, reaction vessel and concentration, can influence the construction of different conformational isomers.Park and co-workers46 have synthesized two conformational isomers (66 and 67) of Ag(I) by changing the reagents' molar ratio (Fig. 22). The 1:1 reaction of AgNO3 and m-bis(cyanobenzyl)sulphide (L31) in a mixture of dichloromethane and methanol leads to the formation of [Ag2(L31)(NO3)2]n (66). However, when the reaction is undertaken in the presence of a large excess of silver(I) nitrate, it generates another isomer, 67. Both isomers feature 2D polymeric arrangements having the same molecular formula. The framework 66 crystallized in the monoclinic P21 space group, whereas 67 crystallized in the monoclinic C2/c space group. The asymmetric unit of 66 contains two Ag(I), one L31 ligand and two NO3− moieties, but in 67, a half molecule of the L31 ligand, one Ag(I) atom and one NO3− ion are present in the asymmetric unit. In both MOFs, the Ag centre has a distorted tetrahedral geometry and is coordinated by one CN nitrogen atom from one m-bis(cyanobenzyl)sulphide ligand, one sulphur atom from a neighbouring m-bis(cyanobenzyl)sulphide ligand and two monodentate NO3− ions. Topological analysis revealed that both frameworks have the same (6,3) network topology.
 | ||
| Fig. 22 Molar ratio-dependent supramolecular isomers 66 and 67 showing the conformational differences of L31 in each compound. Reproduced with permission from ref. 46. Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
The isomerism of these two MOFs arises mainly from the different conformations of the m-bis(cyanobenzyl)sulphide ligand (L31). For 66, the two cyano groups of the ligand are oriented away from each other, forming an “S”-shaped configuration, whereas for 67, the ligand has a “V”-shaped orientation.
Another example concerns the reaction vessel- and concentration-induced conformational supramolecular isomerism, as reported by Wu et al.47 They have synthesized the supramolecular isomers [La2(L32)3(H2O)2(EtOH)(DMF)2]n·3nDMF·3nH2O (68), [La2(L32)3(H2O)4]n·2nH2O (69) and [La2(L32)3(H2O)2(DMF)4]n·2nDMF·2nH2O (70) (L32 = 2,2′-(2,3,5,6-tetramethyl-1,4-phenylene)bis-(methylene)bis(sulfanediyl)dibenzoate) by the solvothermal reaction of H2L32, 1,3-di(pyridin-4-yl)propane and La(NO3)3·6H2O. MOFs 68 and 69 were synthesized under identical reaction conditions except that the reaction vessels were different. The reaction for 68 was carried out in a Teflon-lined steel bomb, whereas 69 was synthesized in a glass tube. On the other hand, 69 and 70 were synthesized under similar conditions, except that the concentrations of the starting reagents were different.
Single crystal X-ray analysis revealed that 68–70 have similar 2D layer-type structures. The asymmetric unit of 68 contains two La ions, three deprotonated L32 ligands, one coordinated ethanol molecule, two coordinated DMF molecules and two coordinated water molecules. However, the asymmetric unit of 69 consists of one La ion, one and half L32 ligands, two coordinated water molecules and one non-coordinated water molecule. In these frameworks, the ligand L32 adopts different syn- and anti-conformations. In 70, the asymmetric unit consists of one La ion, one and half L32 ligands, two coordinated DMF molecules, one coordinated water molecule and one each of non-coordinated water and DMF molecules. In this framework, all the deprotonated ligands adopt the anti-conformation, in contrast to 68 and 69. The conformations of the L32 ligands are highly controlled by the reaction concentration of the starting reagents and reaction vessels that were used during the solvothermal reaction.
4. Catenane supramolecular isomerism
In catenane-based supramolecular isomerism, the MOFs are conformationally and structurally the same but they differ in the level of interpenetration within the structures. Several MOFs having interpenetrated structures have been reported.48 The interpenetration decreases the pore size and volume but it significantly enhances the crystal density and stability of the frameworks. The surface area decreases on account of the interpenetration which may constitute a drawback for the interpenetrated networks, but the change of the pore size can have a significant effect on the guest selectivity. Hence, the structural interpenetration of the frameworks plays an important role in the functional applications of MOFs.However, the synthesis of catenane-based supramolecular isomers in a truly rational manner continues to be a challenging task on account of the many factors that influence the product structures, such as reaction temperature and time, reagent concentration, the presence of template molecules, etc.
4.1 Reagent concentration-dependent catenane isomerism
The first MOFs showing interpenetration/catenation isomers were reported by Yaghi et al.49 who have synthesized four pairs of catenation isomers, 71/71a–74/74a. Isomers 71–74 have doubly interpenetrated structures and were prepared by using a mixture of Zn(NO3)2·4H2O and linear ditopic ligands, i.e. [1,1′-biphenyl]-4,4′-dicarboxylate, 4,5,9,10-tetrahydropyrene-2,7-dicarboxylate, pyrene-2,7-dicarboxylate and [1,1′:4′,1′′-terphenyl]-4,4′′-dicarboxylate (L33), respectively, in N,N-diethylformamide (DEF) under solvothermal conditions. By using more dilute reaction solutions, the noninterpenetrated counterparts 71a–74a were obtained.In this highlight, we have chosen a pair of such isomers as a representative example. [Zn4O(L33)3]n·17nDEF·2nH2O (71a) crystallizes in the cubic Pm3m space group and was synthesized by the reaction of [1,1′:4′,1′′-terphenyl]-4,4′′-dicarboxylic acid and Zn(NO3)2·4H2O in DEF under solvothermal conditions. In this framework, {Zn4O(COO)6} acts as a SBU, being connected to six linear carboxylate ligands forming a 3D cubic net without any interpenetration. However, the doubly interpenetrated [Zn4O(L33)3]n·nDEF·5nH2O (71) crystallizes in the cubic Fm3m space group. It was synthesized under similar conditions but with a higher reagent concentration.
Zaworotko et al. have synthesized two 3D catenation isomers, one being in a noninterpenetrated form, [Cd(bipy)(L34)]n·3nDMF·nH2O (75), and the other having a 2-fold interpenetrated form, [Cd(bipy)(L34)]n (76).50 The solvothermal reaction of 4,4′-bipyridine (bipy), 1,4-benzenedicarboxylic acid (H2L34), and Cd(NO3)2·4H2O in DMF/DEF at 85 °C afforded crystals of framework 75, while performing the similar reaction at a higher temperature (115 °C) led to the formation of 76.
Single crystal X-ray analysis revealed that, in both MOFs, the dinuclear {Cd2N4O8} moiety acts as a SBU, being linked with benzenedicarboxylate (bdc) and creating sheets which are further connected by the 4,4′-bipyridine ligand (Fig. 23A and C). Each Cd(II) ion has a distorted pentagonal bipyramidal geometry with nitrogen atoms from two bipy ligands at the apexes and five carboxylate oxygen atoms at the equatorial positions. The difference between the two frameworks arises from the coordination motifs of the L34 ligand and the twisted nature of the bipy ligand. The cavities within the undulating sheets of 75 and 76 exhibit dimensions of 12.46 × 12.46 Å2 and 10.34 × 15.12 Å2 with corresponding bite angles of 75.9° and 62.6°, respectively.
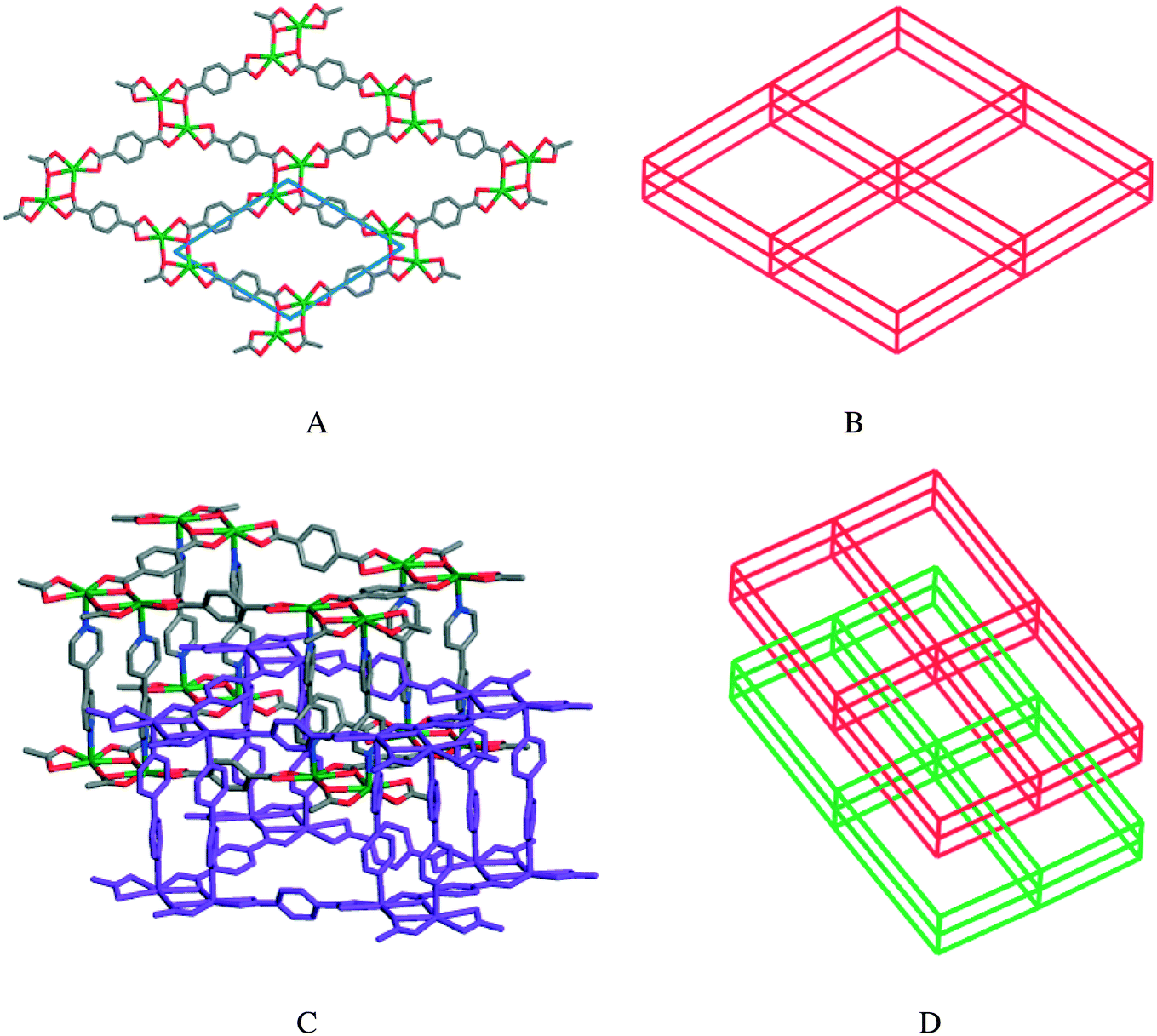 | ||
| Fig. 23 (A) Single [Cd(bdc)] layer in 75 viewed along the c direction. (B) The primitive cubic (pcu) structure of 75. (C) Pillared interpenetrated structure of compound 76. (D) The 2-fold interpenetration in 76. Reprinted with permission from ref. 50. Copyright 2009 American Chemical Society. | ||
A detailed investigation into the effects of the reaction temperature and concentration on the interpenetration showed that a high temperature and a high concentration lead to the interpenetrated MOF (76), whereas the noninterpenetrated framework (75) is formed at lower temperature and concentration. The higher temperatures tend to generate more thermodynamically stable and denser interpenetrated crystals.51,53 However, the lower concentration can reduce the possibility of forming a sublattice in the void of the noninterpenetrated frameworks (Fig. 23B and D).
4.2 Temperature-dependent catenane isomerism
Temperature can have an important influence on the formation of MOF catenane isomers. An example has been described by Xu et al.51 who have synthesized three isomeric MOFs, [Cd(L35)(bipy)]n·4nH2O·2.5nDMF (77), [Cd(L35)(bipy)]n (78) and [Cd(L35)(bipy)]n·4.5nH2O·3nDMF (79) (L35 = 2-amino-1,4-benzenedicarboxylate), by the solvothermal reaction of Cd(NO3)2·4H2O, 4,4′-bipyridyl and 2-amino-1,4-benzenedicarboxylic acid in DMF at different temperatures (Fig. 24). The reaction of the Cd(II) salt with 2-amino-1,4-benzenedicarboxylic acid at 105 °C led to the formation of the noninterpenetrated 3D network 77, whereas upon increasing the reaction temperature to 160 °C the doubly interpenetrated isomer 78 was observed. Moreover, by decreasing the concentrations of the reactants and reaction time, another isomeric framework (79) was prepared.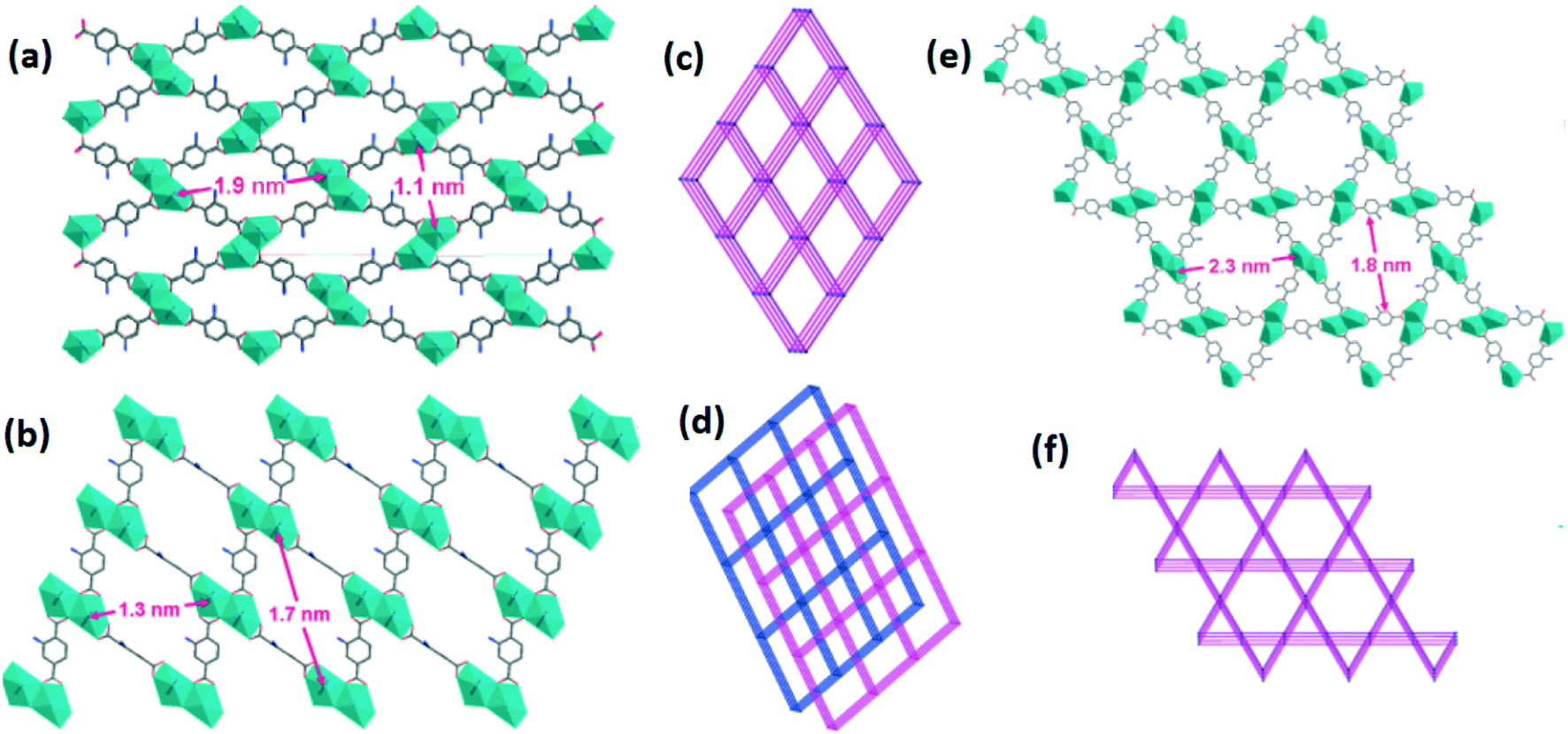 | ||
| Fig. 24 Packing structures of MOFs (a) 77 (prepared at 105 °C), (b) 78 (prepared at 160 °C) and (e) 79 (prepared at 105 °C with different concentrations of the reactants and reaction times); topological diagrams of (c) 77, (d) 78 and (f) 79. Reprinted with permission from ref. 51. Copyright 2010 American Chemical Society. | ||
In compound 77, the asymmetric unit contains one Cd metal ion, one L35 ligand and one bipy. Each Cd(II) ion is seven-coordinated by two nitrogens from two bpy ligands and five carboxylate oxygens from three L35 ligands. The Cd(II) ions are coordinated by the 2-amino-1,4-benzenedicarboxylate ligand forming a layer-type structure which is further connected by bipy ligands generating a 3D pillared-layer structure having a channel size of 1.1 × 1.9 nm2. The asymmetric unit of compound 78 has one Cd ion and two halves of L35 ligands and one bipy. The coordination environments of Cd(II) in 77 and 78 are identical. MOF 75 is a 3D nonporous framework showing 2-fold interpenetration with a planar channel size of 1.3 × 1.7 nm2. This framework has a triangular channel and a larger hexagonal open-channel with a mesoporous size of 1.8 × 2.3 nm2 parallel to the ab plane.
MOFs 77 and 78 are temperature-dependent catenation isomers, whereas they are also conformational isomers due to ca. 90° rotation of one 2-amino-1,4-benzenedicarboxylate ligand. Frameworks 77 and 78 have similar structures, except that in 79 the ligand rotates by ca. 180°. Moreover, 79 undergoes spontaneous transformation to 77 in air upon partial loss of solvents.
Maji et al. reported the catenane supramolecular isomers [Cd(L36)(L37)0.5]n·nDMF·nH2O [L36 = 2-aminobenzenedicarboxylate, L37 = 1,2-bis(4-pyridylmethylene)hydrazine] (80 and 81) which were synthesized under the same molar ratio of organic ligands and metal salts but at different reaction temperatures.52 MOF 80 was prepared at 90 °C, whereas 81 was obtained at a higher temperature (120 °C) (Fig. 25).
 | ||
| Fig. 25 Synthesis and crystal structures of frameworks 80 and 81. Reproduced with permission from ref. 52. Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
X-ray diffraction analysis revealed that both 80 and 81 crystallize in the same crystal system (orthorhombic) but in different space groups (Cmcm for 80 and Cmca for 81). Their asymmetric unit contains one Cd(II) metal ion, one L36 and half L37 ligand along with one each of non-coordinated DMF and H2O molecules. Both frameworks have 2-fold interpenetrated 3D structures. The main difference between them is that in 80 a paddle-wheel {Cd2(COO)4} moiety acts as a SBU, whereas in 81 the SBU concerns the μ-oxo-bridged {Cd2(μ-OCO)2} group. In 80, the Cd(II) ion is five-coordinated, but in 81, it is seven-coordinated. In both MOFs, the metal ion is coordinated by ligand L36 to furnish a 2D square sheet-like structure, being further connected by L37 ligands to extend the networks in three dimensions. Framework 80 has 4 × 4 Å2 square channels but isomer 81 has 3 × 5 Å2 rectangular channels. Both compounds have the same molecular formula but different network topologies involving different SBUs. Also, they are two-fold catenated and can be called as catenane supramolecular isomers.
Gas adsorption analysis of both MOFs revealed an excellent selectivity towards CO2 rather than other gases. They show type II adsorption profiles for N2 adsorption indicating a nonporous nature. However, at 195 K, the first isomer shows an unusual double-step hysteretic CO2 adsorption profile (uptake amount of ca. 118 mL g−1), whereas the second isomer shows a typical type I CO2 profile (uptake amount of 101 mL g−1).
Framework 81 also acts as a very effective catalyst for the Knoevenagel condensation reaction of aldehydes (yield up to 99% within 6 h in acetonitrile, for the case of 4-nitrobenzaldehyde). They also display size selectivity.
Recently, two catenane-based supramolecular isomers of lutetium MOFs, [Lu2(L36)3(H2O)4]n·4nH2O (82) and [Lu2(L36)3(H2O)2(DMF)2]n (83) (L36 = 2-aminoterephthalate), were reported by Gascon et al. (Fig. 26A).53 The reaction of Lu(NO3)3·6H2O with 2-aminoterephthalic acid (H2L36) in water at room temperature led to the formation of the single MOF 82 having a pcu-type net (Fig. 26B). However, performing the reaction at 100 °C in DMF/H2O produced the doubly interpenetrated 3D structure 83 (Fig. 26C).
 | ||
| Fig. 26 A) Synthesis of frameworks 82 and 83. B) The topological representation of the pcu simple net of 82. C) Two-fold interpenetrated network of 83. Reprinted (adapted) with permission from ref. 53. Copyright 2016 American Chemical Society. | ||
Single crystal X-ray diffraction analysis revealed that both MOFs crystallize in the triclinic P space group. The asymmetric unit of 82 contains one lutetium atom, one and a half aminoterephthalate (L36), two coordinated and two non-coordinated water molecules, whereas for 83 the asymmetric unit contains one lutetium atom, one and a half aminoterephthalate (L36) and one each of coordinated water and DMF molecules. For both compounds, the lutetium centre adopts a distorted bicapped trigonal prismatic geometry, surrounded by eight oxygen atoms and forming 3D structures with rhombohedral channels. Topological analysis revealed that both frameworks are a six-connected uninodal net of pcu alpha-Po primitive cubic type with point symbol (412·63).
4.3 Solvent-dependent catenane isomerism
Solvent plays a significant role in the formation of solvent-induced supramolecular isomers and in the crystallization process. Solvent-mediated isomerization can be a recrystallization phenomenon controlled by thermodynamic factors.An example of solvent-induced supramolecular isomerism was reported by Zhou et al.54 who synthesized the MOFs [Cu(L38)(H2O)]n·nDMF·2nH2O (84) and [Cu(L38)(H2O)]n·2nDMF·4.5nH2O (85) (L38 = 3,4′-biphenyldicarboxylate) by solvothermal reactions of the unsymmetrically substituted pro-ligand 3,4′-biphenyldicarboxylic acid (H2L38) with copper nitrate in a mixture of DMF:EtOH:H2O. The use of the 1:1:1 solvent composition led to the formation of 84 but, by changing the solvent ratio to 7:1:1, the isomeric MOF 85 was obtained.
Single-crystal X-ray diffraction revealed that 84 and 85 crystallize in the chiral space group I41 and hexagonal space group R![[3 with combining overline]](https://www.rsc.org/images/entities/char_0033_0305.gif) , respectively. The asymmetric unit of both MOFs contains one Cu(II), one L38 ligand and one coordinated water molecule. The Cu(II) centre has a square pyramidal geometry coordinated by four oxygen atoms from four L38 ligands and one water molecule. Both MOFs contain binuclear paddle-wheel units {Cu2(COO)4} which are connected by the L38 linkers forming 3D frameworks. MOF 84 consists of a 3-fold interpenetrating network, whereas 85 has a 2-fold interpenetrating 3D framework. The topological analysis revealed that 84 has a diamond topology but 85 has a NbO topology.
, respectively. The asymmetric unit of both MOFs contains one Cu(II), one L38 ligand and one coordinated water molecule. The Cu(II) centre has a square pyramidal geometry coordinated by four oxygen atoms from four L38 ligands and one water molecule. Both MOFs contain binuclear paddle-wheel units {Cu2(COO)4} which are connected by the L38 linkers forming 3D frameworks. MOF 84 consists of a 3-fold interpenetrating network, whereas 85 has a 2-fold interpenetrating 3D framework. The topological analysis revealed that 84 has a diamond topology but 85 has a NbO topology.
The N2 and H2 adsorption properties of these MOFs were also tested. The nitrogen sorption isotherm of 84 shows type I sorption behaviour having a BET surface area of 541 m2 g−1, whereas 85 does not show any N2 adsorption. In the case of hydrogen adsorption, frameworks 84 and 85 show H2 uptakes of 2.26 and 1.64 wt%, respectively, at 10 bar.
Vittal and co-workers reported two polyrotaxane-based 2D MOFs with the general formula [Co2(L39)(L40)2]n·nS (86 and 87) [L39 = 1,4-bis(2-(pyridin-4-yl)vinyl)benzene; L40 = sulfonyldibenzoate; S = solvent].55 Framework 86, [Co2(L39)(L40)2]n·1.5nDMA·1.5nH2O (DMA = dimethylacetamide), has a non-interpenetrating structure, but 87, [Co2(L39)(L40)2]n·nDMA·nH2O, is a two-fold interpenetrated 2D framework (Fig. 27). The solvent used in the reaction medium has a strong influence on the formation of these isomers. For example, a mixture of DMA and DMSO led to the formation of compound 86, but addition of water into the reaction mixture generated the isomer 87. The role of water in the formation of 87 is not clear but tentatively can be accounted for by its different polarity (higher than that of DMA or DMSO) which can affect the self-assembly process and lead to the formation of the new network 87. Moreover, addition of water changes the concentration of the reagents and a network with a different interpenetration (87) can be formed.
 | ||
| Fig. 27 Crystal structures of non-interpenetrated framework 86 (A) and doubly interpenetrated framework 87 (B). Reprinted (adapted) with permission from ref. 55. Copyright 2016 American Chemical Society. | ||
Single crystal X-ray diffraction analysis revealed that these MOFs have similar types of connectivity. For both of them, the dinuclear paddle-wheel {Co2(COO)4} unit acts as a building block, being connected by two doubly deprotonated L40 ligands from each side and forming a necklace-like 1D chain, being further connected by the linear spacer ligand L39 to construct a 2D layer structure. The main difference between these frameworks is that 86 has a non-interpenetrated 2D structure, whereas 87 is a 2-fold interpenetrated 2D structure.
A magnetic study of MOF 86 shows antiferromagnetic exchange coupling between the Co(II) ions. When the temperature decreases from 350 K to 30 K, the χmT decreases continuously from 1.47 cm3 K mol−1 to 0.017 cm3 K mol−1 and then remains constant until 2 K.
Another solvent-mediated catenane isomerization was reported by Chung et al.,56 concerning the three isomeric MOFs [Co(L41)2(NCS)2]n·nS (L41 = 1-methyl-10,2-bis(4-pyridyl)ethane; S = guest molecules) (88–90). Layering of a methanolic solution of Co(NCS)2 onto a solution of L41 in CH3NO2 led to the formation of the perpendicularly two-fold interpenetrated 2D grid network 88. Upon keeping the solution for several days, MOF 89 was formed, having mutual interpenetration of two independent 3D CdSO4 networks and 2D grid layers with small voids (Fig. 28). When the diffusion was carried out by layering of a solution of L41 in MeOH onto an aqueous solution of Co(NCS)2 for several days, a noninterpenetrated 2D grid network (90) was obtained. Compounds 88–90 are solvent-mediated catenation isomers.
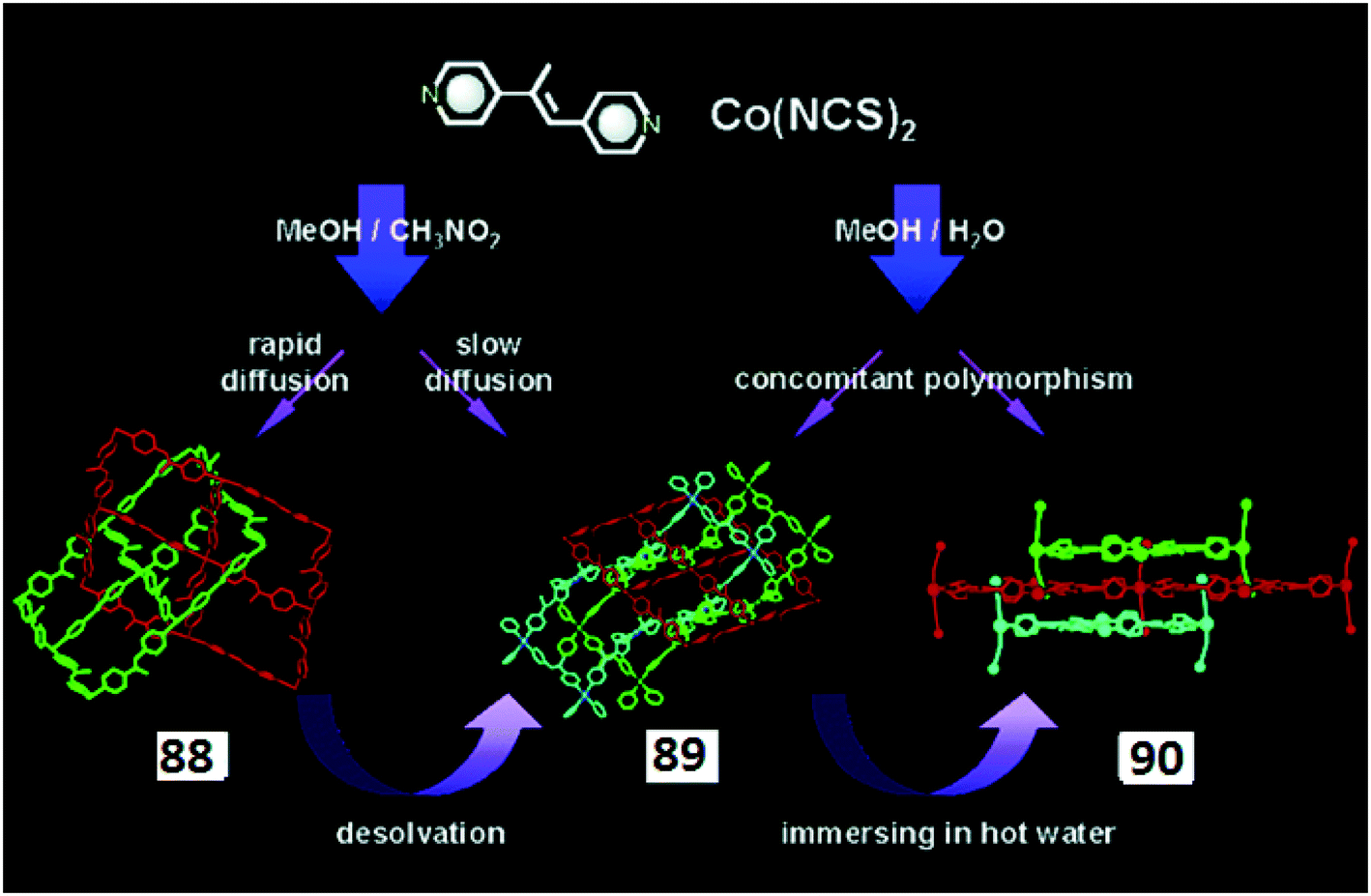 | ||
| Fig. 28 Synthesis, crystal structures and interconversion of compounds 88–90. Reprinted (adapted) with permission from ref. 56. Copyright 2003 American Chemical Society. | ||
4.4 Template or guest molecule-dependent catenane isomerism
The use of appropriate templates or guest molecules during the crystallization process can avoid the interpenetration of the network. Thus, it is reasonable to discuss interpenetrated supramolecular isomers which can be controlled by the use of guest or template molecules.An example, reported by Zhou et al., concerns the pair of catenation isomers [Cu6(L42)4(H2O)6]n·nDMA·12nH2O (91) and [Cu3(L42)2(H2O)3]n·nDMA·2nH2O (92) (L42 = 4,4′,4′′-s-triazine-2,4,6-triyl-tribenzoate).57 The hydrothermal reaction of Cu(NO3)2·2.5H2O and H3L42 in DMA at 75 °C for 48 h led to the formation of the interpenetrated framework 92 (Fig. 29B). But the addition of oxalic acid (as a template) [length of ca. 4.8 Å, calculated by using the Mercury 3.9 crystallographic software] into the same reaction mixture gave the non-interpenetrated network 91 (Fig. 29A). Thus, the catenation of these frameworks is controlled by the presence or absence of template molecules.
 | ||
| Fig. 29 Structure of non-interpenetrated 91 (A) and doubly interpenetrated 92 (B) and the formula of oxalic acid (C). Reprinted with permission from ref. 57. Copyright 2007 American Chemical Society. | ||
Single crystal X-ray diffraction analysis revealed that the MOF 91 crystallized in the cubic Fm3m space group. In this network, the paddle-wheel {Cu2(COO)4} acts as a SBU, being connected through linker L42 and generating a 3D network. Each SBU connects to four L42 ligands and each L42 ligand connects to three SBUs, such assembly leading to the formation of cuboctahedral and octahedral cavities inside the framework. The average pore size of the cuboctahedra is ca. 30 Å and the size of the open square channels is ca. 15.16 and 21.44 Å along the edges or diagonals, respectively.
However, the interpenetrated isomer 92 crystallized in the lower symmetry R3m space group and it is formed from two identical interpenetrated nets. The packing structure of this framework shows that the octahedra of the first network are located inside the cuboctahedral cavities of the other network. The channel size of this framework is reduced due to the interpenetration which is reflected by the solvent-accessible volume of the two frameworks. The solvent-accessible volumes of 87 and 88 are 86% and 74%, respectively (calculated by PLATON).
Both MOFs exhibit permanent porosity and excellent gas adsorption properties. The Langmuir surface areas of 91 and 92 are ca. 2700 m2 g−1 and 3800 m2 g−1, respectively, calculated through the N2 adsorption isotherm. The Langmuir surface area of 92 is higher than that of 91, although the latter has a higher solvent-accessible volume. Hydrogen adsorption studies showed that the activated MOF 91 has a hydrogen uptake of 1.35 wt% at 760 Torr and 77 K, whereas under the same conditions 92 has a significantly higher hydrogen uptake (1.74 wt%). This uncommon phenomenon can be explained by an interaction between the framework walls and gas molecules on account of the development of new adsorption sites in the interpenetrated framework.
MOFs can undergo distortions upon guest removal without changing the metal–ligand connectivities. But a few MOFs can undergo crystal-to-crystal isomerization comprising a metal–ligand connectivity change. For example, the porous metal azolate framework [Ag6Cl(L43)4]n(OH)n·6nH2O (HL43 = 3-amino-1,2,4-triazole) (93) was synthesized by Chen et al. by slow evaporation of an ammonia solution of HL43 and AgCl.58 X-ray diffraction analysis revealed a 2-coordinated Ag(I) connected via a μ3-L43 ligand, generating a 3D structure with five-fold interpenetration. This framework has a large 1D channel along the c-direction with an estimated diameter of ca. 8.5 Å and a solvent-accessible volume of ca. 32.7%. Heating 93 at 375 K for 3 h led to the formation of the crystalline form 94 having a 3D structure with six-fold interpenetration. The solvent-accessible volume of 94 is reduced to 25.2% and the shape of the 1D channel changes to elliptical (4.3 × 10.4 Å2). These two catenation isomers are interconvertible by means of single crystal-to-single crystal transformation (Fig. 30).
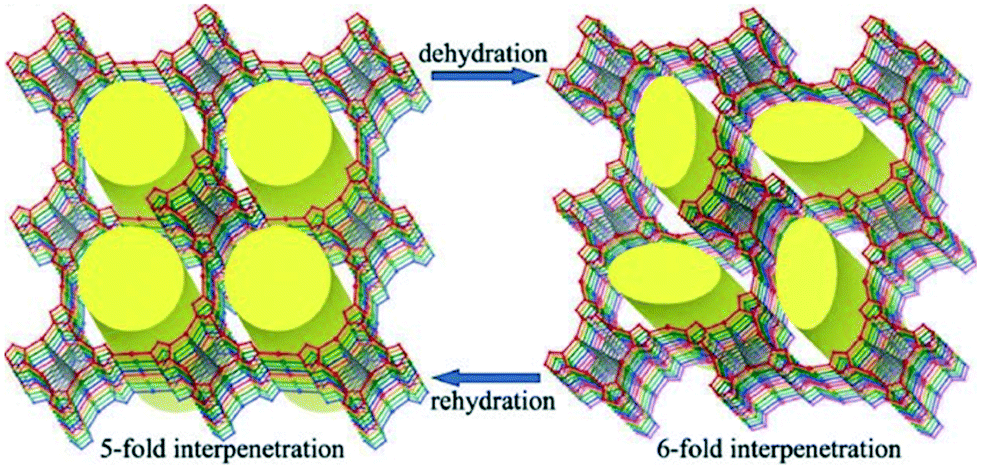 | ||
| Fig. 30 Interconversion of the [Ag6Cl(L43)4] host of 93 and 94 (1D channels are highlighted as yellow columns). Reprinted (adapted) with permission from ref. 58. Copyright 2005 American Chemical Society. | ||
4.5 Catenane isomerism dependent on other factors
Other factors besides those mentioned above can also be responsible for the formation of supramolecular isomers. Su and co-workers have prepared the interpenetrated supramolecular isomers [Cd(L44)2(H2O)]n [L44 = 4-(pyridin-3-ylmethoxy)benzoate] (95–97) by using metal salts with the same metal but containing different anions.59 MOF 95 was formed by the solvothermal reaction of HL44 and Cd(OAc)2·2H2O at pH ≈ 3–4, whereas 96 and 97 were prepared under the same reaction conditions but by using CdCl2 and Cd(NO3)2·2H2O, respectively, instead of Cd(OAc)2·2H2O.The asymmetric unit of these MOFs contains one Cd(II) ion, two L44 ligands and one coordinated water molecule. The Cd(II) ion adopts a pentagonal bipyramidal coordination geometry, surrounded by two N atoms of L44, four carboxylic oxygen atoms of the ligand and one water molecule. The single crystal X-ray diffraction analysis revealed that 95 has a 2-fold parallel interpenetrated 2D network structure, whereas 96 has a single (44,62) 2D network and 97 has a 2D structure with a (66) topological net. Compounds 95–97 can be considered as catenation supramolecular isomers.
Ahn et al. reported a sonochemical method to achieve control of catenation in MOFs.60 They have synthesized the two catenation isomers [Cu6(L45)4(H2O)6]n·nDMA·12nH2O (98) and [Cu3(L45)2(H2O)3]n·nDMA·2nH2O (99) (L45 = 4,4′,4′′-s-triazine-2,4,6-triyl-tribenzoate) by using a sonochemical method for 1 h at 20 kHz. A low sonication power (150 W) led to the non-interpenetrated framework 98, but at high sonication levels (300 W) the production of the interpenetrated product 99 is promoted (Fig. 31). The CO2 adsorption properties of these MOFs were studied at 273 and 298 K. The catenated framework 99 shows a higher CO2 adsorption capacity (189 mg g−1) than the non-catenated (98) one (156 mg g−1) at 298 K. 99 also displays a higher gas adsorption capacity (1171 mgCO2 gadsorbent−1) at 30 bar and 298 K.
 | ||
| Fig. 31 The sonochemical preparation of frameworks 98 and 99 is schematically shown. Reprinted (adapted) with permission from ref. 60. Copyright 2011 American Chemical Society. | ||
5. Optical supramolecular isomerism
Optical isomers are a subject of high interest due to their structures and enormous potential applications, namely in molecular recognition, non-linear optical materials, asymmetric catalysis, enantiomeric separation, etc.61 One of the possible ways of generating optical isomers is through the employment of chiral ligands, which can transfer their chirality to the metal centre(s), eventually forming isomeric structures in a stereocontrolled way. MOFs concern usually complicated systems and optical supramolecular isomers therein are less common than molecular optical isomers.An example of optical isomers in Ag(I) MOFs was reported by Shen et al.62 who have synthesized two pairs of enantiomers, namely R-[Ag4(L46)4]n·3nDMA (100), S-[Ag4(L46)4]n·3nDMA (101), R-[Ag(L46)]n (102) and S-[Ag(L46)]n (103). Compounds 100 and 101 were obtained by the reaction of R- or S-HL46 [HL46 = 2-(1-hydroxyethyl) benzimidazole] with AgNO3 in aqueous ammonia and dimethylacetamide (DMA) (Fig. 32). However, MOFs 102 and 103 were obtained through a similar procedure except by using methanol instead of DMA as solvent.
 | ||
| Fig. 32 (a) The structure of HL46; (b) 1D linear chain of compounds 100 and 101. (c) Views of space-filling models of 100 (left) and 101 (right). (d) Space-filling representation of the left-handed helix in 102 (left) and the right-handed helix in 103 (right); (e) top view of the pseudo-121 helix in 102 (left) and 103 (right) along the c-axis. Reprinted (adapted) with permission from ref. 61. Copyright 2014 Elsevier. | ||
Single crystal structural analyses revealed that MOFs 100 and 101 are a pair of optical enantiomers and display a 1D linear chain. They crystallized in the triclinic P space group with similar asymmetric units containing two Ag(I) atoms, two L46 ligands and 1.5 non-coordinated DMA solvent molecules. In these frameworks, four symmetrically non-equivalent molecules (Z′ = 4) are present in the lattice. Each Ag(I) centre is bi-coordinated via two N-atoms from two L42 ligands, forming a 1D linear chain of Ag and L42.
The other pair of optical enantiomers (102 and 103) crystallize in a hexagonal crystal system but in different space groups, P6522 (for 102) and P6122 (for 103). They have similar asymmetric units which contain two Ag(I) atoms with half occupancy and one L46 ligand. Each Ag atom is coordinated by two N-atoms from two neighbouring L46 ligands and the ligands are coordinated in cis (for 102) or trans (for 103) fashion and create an arc fragment. These arc fragments interconnect via Ag ions into a single-stranded left-handed or right-handed helix along the c-axis for 102 or 103, respectively. The results show that optical isomerism of homochiral metal benzimidazolate MOFs can be prompted by the solvent.
The solid-state circular dichroism (CD) spectra were measured from KBr pellets for MOFs 100–103. The opposite signs of Cotton effects further confirm that compounds 100 and 101 as well as 102 and 103 constitute two pairs of enantiomers, with the optical activities originating from the chiral ligand.
Chiral MOFs can be obtained by applying chiral ligands, as discussed above, but cases are also known63 of optical isomers which are prepared from achiral ligands as illustrated below.
Bu et al. have reported64 a pair of 3D enantiomorphic frameworks, formulated as [Mn(L47)2]n (104 and 105) [HL47 = N-(1H-tetrazol-5-yl)benzamide], synthesized by the reaction of HL47 and MnCl2·4H2O. During the crystallization process, spontaneous resolution occurred and two different crystals with chiral space P41212 for 104 and P43212 for 105 were obtained (Fig. 33A and B).
 | ||
| Fig. 33 Crystal structures of 104 (A) and 105 (B); (C) molecular structure of HL47; (D) the enantiomeric nature of 104 and 105. Reprinted (adapted) with permission from ref. 64. Copyright 2010 Royal Society of Chemistry. | ||
Both frameworks have similar asymmetric units that contain one Mn(II) ion and one deprotonated L47. The Mn(II) centre shows a slightly distorted octahedral geometry and is coordinated by four N atoms from four different L47 ligands and two O atoms from two L47 ligands. This ligand acts as a tridentate ligand and coordinates to three Mn(II) ions simultaneously (two via N atoms of the tetrazole ring and one carbonyl O atom). The distances between adjacent Mn(II) centres are 6.741 Å (for 104) and 6.714 Å (for 105). Topological analysis revealed that both frameworks have the same dia topology. In these MOFs, the chirality arises mainly from the screw coordination arrangement of the achiral ligand (L47) around the Mn(II) centres. Moreover, compounds 104 and 105 are enantiomeric with each other as reflected by the mirror structures (Fig. 33D). The CD spectra of 104 and 105 confirm the formation of the pair of enantiomeric frameworks. The low-temperature magnetic study of these species shows antiferromagnetic interactions between Mn(II) ions.
Another example of chiral MOFs derived from achiral ligands by spontaneous resolution was reported by Yan and co-workers.65 They have synthesized the pseudopolymorphous [Mn2(N3)4(L48)]n (106) and [Mn2(N3)4(L48)]n·nMeOH (107) [L48 = 2,5-bis(2-pyridyl)-3,4-diaza-2,4-hexadiene] by the reaction of Mn(ClO4)2·6H2O, L48 and NaN3 in methanol at room temperature. 106 and 107 crystallized with a prismatic and a hexagonal shape, respectively. The single crystal X-ray analyses revealed that both crystals have a 2D structure but 106 crystallizes in a centrosymmetric space group (C2/c) and 107 in a chiral one (P3121). Both structures consist of homochiral 2D layers with the same topology and similar structural parameters. Each Mn(II) centre is octahedrally coordinated by two N atoms from one L48 ligand and four azide ligands. The low-temperature magnetic study shows that both frameworks are weak ferromagnets with different critical temperatures (8 K for 106 and 12.5 K for 107), whose spontaneous magnetization arises from spin canting within the antiferromagnetic layers (canted antiferromagnetism), consistent with the chiral layer structure.
Ghosh et al. reported66 the homochiral framework [Zn(L49)(H2O)2]n·2nNO3·xnG (108) (G = disordered solvent) synthesized from the achiral ligand 4,4′-(ethane-1,2-diyl)bis(N-(pyridin-2-ylmethylene)aniline) (L49) and Zn(NO3)2 in a solvent mixture of CH2Cl2/EtOH, H2O and benzene. They have prepared another isomeric chiral framework [Zn(L49)(H2O)2]n·2nClO4·2nH2O (109) by keeping 108 in an aqueous solution of NaClO4 for ca. 5 days.
Single-crystal X-ray diffraction analysis revealed that both frameworks crystallized in a chiral hexagonal crystal system but in different space groups, P6522 (for 108) and P65 (for 109). Both MOFs have similar asymmetric units which contain one-half Zn(II) ion, one-half L49, one coordinated water, two nitrate or perchlorate counter-ions (with half occupancy each) and solvents. Each zinc centre is octahedrally coordinated by four nitrogen atoms from two different L49 ligands and two oxygen atoms from two water molecules. The ligand L49 coordinates to the metal ion in such a way that it gives a helical arrangement to the structure. Extending the coordination at both ends of the ligand furnishes an infinite 1D helical chain with pitches of 44.042 Å (for 108) and 43.105 Å (for 109). The overall framework shows a six-fold interwoven helical packing. Solid-state CD spectral analysis revealed that the overall frameworks are homochiral in nature.66
Conclusions
Supramolecular isomerism in the area of metal organic frameworks (MOFs) or coordination polymers (CPs) is still an underdeveloped field of research on account of the difficulties associated with the rational design, synthesis and prediction of the structure and properties of such frameworks.In this highlight, we attempt to gather illustrative examples of supramolecular isomers of MOFs, grouped in four main categories, i.e., structural, conformational, catenane and optical isomers. Although structural and catenane isomers are already well documented, only a few examples of conformational isomers have been reported, whereas optical isomers are even rarer.
The construction of isomeric MOFs is challenging, being dependent on many factors, and thus their synthetic routes are addressed, herein, showing the relevance of key factors, such as the reaction temperature, reagent concentration, solvent, additive, template/guest, crystallization methods, etc., which can determine the types of obtained isomers.
Examples of (designed) supramolecular isomerism are discussed, representing steps towards the design and control of the structures of MOF isomers. The field is expected to undergo extensive developments, and continued efforts are required to generalize rational approaches to the synthesis of targeted MOFs with required properties. Supramolecular isomerism in MOFs provides a good opportunity for investigation of structure–property relationships with potential application, e.g., in the construction of functional materials, and deserves to be further explored.
Acknowledgements
This work has been supported by the Foundation for Science and Technology (FCT), Portugal (project UID/QUI/00100/2013). Authors A. Karmakar and A. Paul express their gratitude to the FCT for post-doctoral fellowships (ref. no. SFRH/BPD/76192/2011 and SFRH/BPD/88450/2012).References
-
(a) Z. Zhang and M. J. Zaworotko, Chem. Soc. Rev., 2014, 43, 5444–5455 RSC
; (b) V. Guillerm, D. Kim, J. F. Eubank, R. Luebke, X. Liu, K. Adil, M. S. Lah and M. Eddaoudi, Chem. Soc. Rev., 2014, 43, 6141–6172 RSC
-
(a) J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC
-
(a) Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC
-
(a) Y. He, W. Zhou, G. Qian and B. Chen, Chem. Soc. Rev., 2014, 43, 5657–5678 RSC
-
(a) M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353–1379 RSC
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC
-
(a) H.-C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC
-
(a) A. Karmakar, R. J. Sarma and J. B. Baruah, Eur. J. Inorg. Chem., 2007, 643–647 CrossRef CAS
- B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS PubMed
- J.-P. Zhang, X.-C. Huang and X.-M. Chen, Chem. Soc. Rev., 2009, 38, 2385–2396 RSC
- T. A. Makal, A. A. Yakovenko and H.-C. Zhou, J. Phys. Chem. Lett., 2011, 2, 1682–1689 CrossRef CAS
-
(a) A. J. Blake, N. R. Brooks, N. R. Champness, M. Crew, A. Deveson, D. Fenske, D. H. Gregory, L. R. Hanton, P. Hubberstey and M. Schroder, Chem. Commun., 2001, 1432–1433 RSC
- I.-H. Park, R. Medishetty, J.-Y. Kim, S. S. Lee and J. J. Vittal, Angew. Chem., Int. Ed., 2014, 53, 1–6 CrossRef
-
(a) N. Zhu, M. J. Lennox, T. Dürenb and W. Schmitt, Chem. Commun., 2014, 50, 4207–4210 RSC
-
(a) W.-W. Dong, D.-S. Li, J. Zhao, L.-F. Ma, Y.-P. Wu and Y.-P. Duan, CrystEngComm, 2013, 15, 5412–5416 RSC
- X. Zhao, Nano Reports, 2015, 1, 42–50 Search PubMed
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC
- M.-A. Munoz-Hernandez, T. S. Keizer, P. Wei, S. Parkin and D. A. Atwood, Inorg. Chem., 2001, 40, 6782–6787 CrossRef CAS PubMed
- A. Karmakar, G. M. D. M. Rúbio, M. F. C. Guedes da Silva, S. Hazra and A. J. L. Pombeiro, Cryst. Growth Des., 2015, 15, 4185–4197 CAS
- A. Karmakar, M. F. C. Guedes da Silva, S. Hazra and A. J. L. Pombeiro, New J. Chem., 2015, 39, 3004–3014 RSC
- C.-P. Li, J. Chen, P.-W. Liu and M. Du, CrystEngComm, 2013, 15, 9713–9721 RSC
- T. Panda, T. Kundu and R. Banerjee, Chem. Commun., 2013, 49, 6197–6199 RSC
- X.-G. Guo, W.-B. Yang, X.-Y. Wu, Q.-K. Zhang, L. Lin, R. M. Yu and C.-Z. Lu, CrystEngComm, 2013, 15, 3654–3663 RSC
- G.-D. Zou, Z.-Z. He, C.-B. Tian, L.-J. Zhou, M.-L. Feng, X.-D. Zhang and X.-Y. Huang, Cryst. Growth Des., 2014, 14, 4430–4438 CAS
- X.-C. Huang, D. Li and X.-M. Chen, CrystEngComm, 2006, 8, 351–355 RSC
- S. Hazra, M. F. C. Guedes da Silva, A. Karmakar and A. J. L. Pombeiro, RSC Adv., 2015, 5, 28070–28079 RSC
- P. Kanoo, K. L. Gurunatha and T. K. Maji, Cryst. Growth Des., 2009, 9, 4147–4156 CAS
- A. Mahmoudi, S. Dehghanpour, C. Gholamrezazadeh, M. Jahanbakhshyan, A. H. Mahmoudkhani and N. Attari, Polyhedron, 2012, 42, 265–270 CrossRef CAS
- Z.-G. Gu, G.-Z. Li, P.-Y. Yin, Y.-N. Chen, H.-M. Peng, M.-F. Wang, F. Cheng, F.-L. Gu, W.-S. Li and Y.-P. Cai, Inorg. Chem. Commun., 2011, 14, 1479–1484 CrossRef CAS
- S. Masaoka, D. Tanaka, Y. Nakanishi and S. Kitagawa, Angew. Chem., Int. Ed., 2004, 43, 2530–2534 CrossRef CAS PubMed
- S. S. Nagarkar, A. K. Chaudhari and S. K. Ghosh, Cryst. Growth Des., 2012, 12, 572–576 CAS
- L. Fan, X. Zhang, Z. Sun, W. Zhang, Y. Ding, W. Fan, L. Sun, X. Zhao and H. Lei, Cryst. Growth Des., 2013, 13, 2462–2475 CAS
- Y.-L. Bai, J. Tao, R.-B. Huang and L.-S. Zheng, CrystEngComm, 2008, 10, 472–474 RSC
- F. Dai, H. He and D. Sun, Inorg. Chem., 2009, 48, 4613–4615 CrossRef CAS PubMed
- T. Panda, P. Pachfule and R. Banerjee, Chem. Commun., 2011, 47, 7674–7676 RSC
-
(a) S. Amirjalayer and R. Schmid, J. Phys. Chem. C, 2008, 112, 14980–14987 CrossRef CAS
- B. Chakraborty, P. Halder and T. K. Paine, Dalton Trans., 2011, 40, 3647–3654 RSC
-
(a) I.-H. Park, S. S. Lee and J. J. Vittal, Chem. – Eur. J., 2013, 19, 2695–2702 CrossRef CAS PubMed
- D. Sun, Y. Ke, T. M. Mattox, B. A. Ooro and H.-C. Zhou, Chem. Commun., 2005, 5447–5449 RSC
- X.-S. Wang, S. Ma, P. M. Forster, D. Yuan, J. Eckert, J. J. López, B. J. Murphy, J. B. Parise and H.-C. Zhou, Angew. Chem., Int. Ed., 2008, 47, 7263–7266 CrossRef CAS PubMed
- C. Serre, F. Millange, C. Thouvenot, M. Noguès, G. Marsolier, D. Louër and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519–13526 CrossRef CAS PubMed
- D.-S. Chen, L.-B. Sun, Z.-Q. Liang, K.-Z. Shao, C.-G. Wang, Z.-M. Su and H.-Z. Xing, Cryst. Growth Des., 2013, 13, 4092–4099 CAS
-
(a) W.-C. Song, X.-Z. Cui, Z.-Y. Liu, E.-C. Yang and X.-J. Zhao, Sci. Rep., 2016, 6, 34870 CrossRef CAS PubMed
- C.-P. Li, J.-M. Wu and M. Du, Inorg. Chem., 2011, 50, 9284–9289 CrossRef CAS PubMed
- R. Ding, C. Huang, J. Lu, J. Wang, C. Song, J. Wu, H. Hou and Y. Fan, Inorg. Chem., 2015, 54, 1405–1413 CrossRef CAS PubMed
- E. Lee, J.-Y. Kim, S. Sung Lee and K.-M. Park, Chem. – Eur. J., 2013, 19, 13638–13645 CrossRef CAS PubMed
- P. Cui, J. Dou, D. Sun, F. Dai, S. Wang, D. Sun and Q. Wu, CrystEngComm, 2011, 13, 6968–6971 RSC
- Y. He, B. Li, M. O'Keeffe and B. Chen, Chem. Soc. Rev., 2014, 43, 5618–5656 RSC
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed
- J. J. Zhang, L. Wojtas, R. W. Larsen, M. Eddaoudi and M. J. Zaworotko, J. Am. Chem. Soc., 2009, 131, 17040–17041 CrossRef CAS PubMed
- H. L. Jiang, Y. Tatsu, Z. H. Lu and Q. Xu, J. Am. Chem. Soc., 2010, 132, 5586–5587 CrossRef CAS PubMed
- R. Haldar, S. K. Reddy, V. M. Suresh, S. Mohapatra, S. Balasubramanian and T. K. Maji, Chem. – Eur. J., 2014, 20, 4347–4356 CrossRef CAS PubMed
- A. Dikhtiarenko, P. Serra-Crespo, S. Castellanos, A. Pustovarenko, R. Mendoza-Meroño, S. García-Granda and J. Gascon, Cryst. Growth Des., 2016, 16, 5636–5645 CAS
- L. Feng, Z. Chen, T. Liao, P. Li, Y. Jia, X. Liu, Y. Yang and Y. Zhou, Cryst. Growth Des., 2009, 9, 1505–1510 CAS
- I.-H. Park, H. Ju, T. S. Herng, Y. Kang, S. S. Lee, J. Ding and J. J. Vittal, Cryst. Growth Des., 2016, 16, 7278–7285 CAS
- D. M. Shin, I. S. Lee, D. Cho and Y. K. Chung, Inorg. Chem., 2003, 42, 7722–7724 CrossRef CAS PubMed
- S. Ma, D. Sun, M. Ambrogio, J. A. Fillinger, S. Parkin and H. C. Zhou, J. Am. Chem. Soc., 2007, 129, 1858–1859 CrossRef CAS PubMed
- J.-P. Zhang, Y.-Y. Lin, W.-X. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2005, 127, 14162–14163 CrossRef CAS PubMed
- S.-L. Li, K. Tan, Y.-Q. Lan, J.-S. Qin, M.-N. Li, D.-Y. Du, H.-Y. Zang and Z.-M. Su, Cryst. Growth Des., 2010, 10, 1699–1705 CAS
- J. Kim, S.-T. Yang, S. B. Choi, J. Sim, J. Kim and W.-S. Ahn, J. Mater. Chem., 2011, 21, 3070–3076 RSC
-
(a) T. Nakano and Y. Okamoto, Chem. Rev., 2001, 101, 4013–4038 CrossRef CAS PubMed
- X.-G. Liu, Y. Y. Wang, Y.-Y. Hu, Z.-G. Guc and L. Shen, Inorg. Chem. Commun., 2014, 40, 103–107 CrossRef CAS
-
(a) Y. Q. Lan, L. S. Li, X. L. Wang, K. Z. Shao, D. Y. Du, Z. M. Su and E. B. Wang, Chem. – Eur. J., 2008, 14, 9999–10006 CrossRef CAS PubMed
- X.-L. Tong, T.-L. Hu, J.-P. Zhao, Y.-K. Wang, H. Zhang and X.-H. Bu, Chem. Commun., 2010, 46, 8543–8545 RSC
- E.-Q. Gao, Y.-F. Yue, S.-Q. Bai, Z. He and C.-H. Yan, J. Am. Chem. Soc., 2004, 126, 1419–1429 CrossRef CAS PubMed
- B. Manna, B. Joarder, A. V. Desai, A. Karmakar and S. K. Ghosh, Chem. – Eur. J., 2014, 20, 12399–12404 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2017 |