
Experimental and theoretical investigations of the gas adsorption sites in rht-metal–organic frameworks
Tony
Pham
 *,
Katherine A.
Forrest
,
Douglas M.
Franz
and
Brian
Space
*
*,
Katherine A.
Forrest
,
Douglas M.
Franz
and
Brian
Space
*
Department of Chemistry, University of South Florida, 4202 East Fowler Avenue, CHE205, Tampa, FL 33620-5250, USA. E-mail: tpham4@mail.usf.edu; brian.b.space@gmail.com
First published on 20th July 2017
Abstract
rht-metal–organic frameworks (MOFs) represent a highly popular class of MOFs in the world of porous crystalline materials. MOFs belonging to this family consist of M2+ ions coordinated to hexatopic organic linkers containing three coplanar isophthalate-based moieties. rht-MOFs are a promising platform of MOFs because they display open-metal sites through the [M2(O2CR)4] clusters, high surface areas, and tunable pore sizes and chemical functionalities. They have been shown to exhibit high uptake for various energy-related gases, such as H2 and CO2. Detailed insights into the gas sorption mechanisms and binding sites in these MOFs can be made by way of experimental techniques, including neutron powder diffraction (NPD) and inelastic neutron scattering (INS), and theoretical methods, such as Monte Carlo (MC) simulations and electronic structure calculations. In this highlight, we review the important experimental and theoretical studies that have been performed to investigate the favorable gas sorption sites in these MOFs. A better understanding of the gas sorption mechanisms in rht-MOFs and related structures can allow for the rational design of new materials that are tailored for specific applications.
 Tony Pham | Tony Pham was born and raised in St. Petersburg, Florida. He obtained his B.S. degree at the University of South Florida (USF) in 2008. In 2015, he received his Ph.D. in Computational Chemistry at USF working under the advisement of Professor Brian Space. His research involves the modeling of gas adsorption and separation in various porous materials, including metal–organic frameworks (MOFs). As a Postdoctoral Researcher at USF, he remains working closely with Dr. Space’s research group and experimental collaborators to elucidate the gas adsorption mechanisms in MOFs using sophisticated modeling techniques. |
 Katherine A. Forrest | Katherine A. Forrest was born in San Francisco, California and raised in Clearwater, Florida. She received a B.S. degree in Chemistry at the University of South Florida (USF) in 2008. As a Ph.D. candidate, she works under Professor Brian Space performing research in the field of computational chemistry at USF. The main focus of her current research regards metal–organic frameworks (MOFs) for gas storage, separations, and catalysis. Specifically, her work involves the implementation of computational methods to understand the atomistic behavior in MOFs in collaboration with experimental chemists. |
 Douglas M. Franz | Douglas M. Franz was born in Seminole, Florida and received a B.S. degree in Environmental Science & Policy from the University of South Florida (USF) in 2013. He is now a Ph.D. candidate working under Professor Brian Space, with a research focus on computer simulation of guest molecules in crystalline materials, which have applications in gas adsorption, separation, and catalysis. He actively contributes toward both the application and developmental aspects of Dr. Space’s research group in efforts to gain quantitative insight on submicroscopic phenomena, such as adsorption, selectivity, and diffusion of gases in metal–organic frameworks (MOFs). |
 Brian Space | Brian Space obtained his Ph.D. in 1992 from Boston University under the supervision of Professor David Coker. Dr. Space is an NSF Career Award winner who, after appointments at Duquesne and Princeton University, joined the University of South Florida (USF) in Tampa, where he is currently a Professor of Chemistry. His research group is primarily concerned with computer simulation of condensed phase phenomena. Current focus is the development of highly accurate potential energy functions for environmentally relevant gases, which are then employed in molecular simulations of gas adsorption within metal–organic frameworks (MOFs). |
I. Introduction
Metal–organic frameworks (MOFs) are solid crystalline materials that are synthesized from metal ions and organic ligands (or “linkers”).1 These two components combine together in a self-assembly process to create a three-dimensional framework that is often porous. The pores of these materials can be used to sorb guest molecules, such as H2, CO2, CH4, and N2. Because of their porous nature, MOFs have been considered to be promising candidates for a variety of gas sorption applications, such as H2 storage,2 CH4 storage,3 and CO2 capture and sequestration.4 MOFs have the ability to capture a large amount of sorbate molecules within their pores and release them freely through adjustments in thermodynamic conditions. MOFs are highly tunable, as a number of different structures can be synthesized by changing the metal ion and/or organic ligand.5 Indeed, over 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 different MOF structures with distinct pore sizes, topologies, and chemical functionalities have been synthesized and reported in the literature.6
000 different MOF structures with distinct pore sizes, topologies, and chemical functionalities have been synthesized and reported in the literature.6
The rht-MOF platform is a highly popular class of MOFs that exists in the literature.7–40 These MOFs are synthesized by combining metal ions in the 2+ oxidation state with hexatopic ligands that contain three coplanar isophthalate-based moieties. Each carboxylate group of the ligand coordinates to M2+ ions (usually Cu2+) to form the square metal paddlewheel, [M2(O2CR)4], molecular building block (MBB). A total of 48 carboxylate groups are connected to M2+ ions to form 12 paddlewheel MBBs; these MBBs are connected through the isophthalate-based components of the ligand to give rise to a cuboctahedron (cub-Oh). The center of the ligand, which typically constitutes a component with C3 symmetry, represents a triangular building block. The points of extension from the isophthalate-based group correspond to an edge of the cub-Oh. There are 24 edges of a cub-Oh and each edge is connected to triangular building blocks (i.e., the central components of the triisophthalate-based ligands) to give the (3,24)-connected rht net. The overall structure of rht-MOFs consists of three distinct porous cages: the cub-Oh, the truncated tetrahedron (T-Td), and the truncated octahedron (T-Oh). A schematic of how rht-MOFs are constructed is shown in Fig. 1. Over 40 different rht-MOF structures have been synthesized and reported in the literature. rht-MOFs are a promising class of MOFs due to the presence of open-metal sites (also known as unsaturated metal centers), high surface areas, and tunable pore sizes and functionalities. They have been shown to display high uptake for a number of energy-related gases.
 | ||
| Fig. 1 A scheme showing the construction of rht-MOFs using rht-MOF-1 as an example. The isophthalate (or isophthalate-based) groups (line) combine with metal ions through the oxygen atoms to form the [M2(O2CR)4] MBBs (square). These components combine together to form a cub-Oh. The point of extension from each isophthalate component (i.e., each edge of the cub-Oh) is connected to a component that has C3 symmetry (triangle). The resulting topology of rht-MOFs consists of three distinct cages: cub-Oh (green), T-Td (blue), and T-Oh (yellow). This figure was reproduced from ref. 25 within the guidelines provided by Wiley-VCH (Germany). Copyright 2012 Wiley-VCH (Germany). | ||
In addition to experimental gas sorption measurements, various experimental and theoretical studies have been performed to pinpoint the binding sites and elucidate the gas sorption mechanism in rht-MOFs. In general, the sorption process and nature of the binding sites in MOFs are often not well-understood. Experimental techniques to probe the binding sites in such materials typically include neutron powder diffraction (NPD) or inelastic neutron scattering (INS), while theoretical methods involve grand canonical Monte Carlo (GCMC) simulations or electronic structure calculations. In this highlight, we provide an overview of the significant experimental and theoretical studies that have been executed to investigate the sorption sites in different members of the popular rht-MOF platform. These studies have yielded detailed insights into the gas sorption mechanism in this class of MOFs. While this paper aims to survey a large amount of research, we apologize to any authors whose work has been omitted in this highlight due to oversight.
II. Experimental studies
A. Neutron powder diffraction
To the best of our knowledge, the first important experimental study that yielded insights into the binding sites in an rht-MOF was performed by Yan et al. in 2010, where the authors carried out NPD experiments of D2 sorbed in NOTT-112.13 NOTT-112 is an rht-MOF that consists of Cu2+ ions coordinated to 1,3,5-tris(3′,5′-dicarboxy[1,1′-biphenyl]-4-yl)benzene linkers (L4 in Fig. 2).9 Note, an organic representation of the linkers used to synthesize all rht-MOFs discussed in this highlight are listed in Fig. 2. In addition, the experimental properties and selected gas sorption data for these MOFs are summarized in Tables 1 and 2, respectively. NOTT-112 was shown to have a Brunauer–Emmett–Teller (BET) surface area41 of 3800 m2 g−1 and a pore volume within the range of 1.6–1.7 cm3 g−1.9 Further, this MOF displayed an absolute H2 uptake of 2.3 wt% at 78 K/1 bar and a zero-loading isosteric heat of adsorption (Qst) value of 5.64 kJ mol−1 according to experimental measurements. The D2 sorption sites observed in the MOF from NPD studies are shown in Fig. 3. | ||
| Fig. 2 Organic representations of the linkers (L1–L9) used to synthesize the rht-MOFs discussed in this highlight. The associated names of the rht-MOFs are summarized in Table 1. | ||
| Name | Ligandd | S L (m2 g−1) | S BET (m2 g−1) | V p (cm3 g−1) | Density (g cm−3) | Reference |
|---|---|---|---|---|---|---|
| a Also known as Cu-TDPAT. b Also known as NOTT-122a or NU-125. c Also known as Cu-TDPAH. d Organic representations of the ligands are depicted in Fig. 2. SL = Langmuir surface area. SBET = BET surface area. Vp = Pore volume. | ||||||
| rht-MOF-1 | L1 | 3223 | 2847 | 1.01 | 0.702 | 7 |
| rht-MOF-pyr | L2 | — | 2133 | — | 0.708 | 35 |
| PCN-61 | L3 | 3500 | 3000 (3350) | 1.36 (1.37) | 0.56 | 12 (15) |
| NOTT-112 | L4 | — | 3800 | 1.62 | 0.503 | 9 |
| Cu-TPBTM | L5 | 3570 | 3160 | 1.27 | 0.627 | 15 |
| rht-MOF-4a | L6 | 1590 | 1070 | 1.10 | 0.665 | 25 |
| rht-MOF-7a | L7 | 2608/2170 | 1938/— | 0.93/0.76 | 0.782/0.788 | 19/20 |
| NTU-105b | L8 | — | 3543/3286/3120 | 1.33/1.41/1.29 | 0.598/0.589/0.578 | 27/28/29 |
| rht-MOF-9c | L9 | 3070/2540 | 2420/2171 | 0.943/0.91 | 0.742/0.89 | 31/33 |
| Name | H2 uptaked (wt%) | H2Qste (kJ mol−1) | CO2 uptakef (mmol g−1) | CO2Qste (kJ mol−1) | CH4 uptakef (mmol g−1) | CH4Qste (kJ mol−1) | Reference |
|---|---|---|---|---|---|---|---|
| a Also known as Cu-TDPAT. b Also known as NOTT-122a or NU-125. c Also known as Cu-TDPAH. d At 77 K/1 atm. e At the lowest loading evaluated. f At 298 K/1 atm. | |||||||
| rht-MOF-1 | 2.4 (—/1.81) | 9.5 | (2.57/2.43) | (32.5/29.0) | (—/0.46) | (—/19.3) | 7 (20/36) |
| rht-MOF-pyr | 1.74 | — | 2.96 | 27.7 | 0.50 | 14.2 | 36 |
| PCN-61 | 2.25 | 6.36 | (3.15) | 21.0 (22.0) | 0.79 | 13.79 | 12 (15) |
| NOTT-112 | 2.3 | 5.64 | — | — | — | — | 9 |
| Cu-TPBTM | (2.61) | (6.6) | 5.29 | 26.3 | — | — | 15 (58) |
| rht-MOF-4a | 1.9 | 9.5 | — | — | — | — | 25 |
| rht-MOF-7a | 2.65/—(2.20) | 8.29/—(6.77) | 5.9/3.9 | 42.2/44.7 | 1.1/— | — | 19/20 (48) |
| NTU-105b | 2.75/2.6/2.4 | 6.61/6.0/5.2 | 4.2/4.63/4.0 | 36/24.5/23.8 | —/0.82/0.83 | —/—/15.5 | 27/28/29 |
| rht-MOF-9c | 2.72/— | 6.9/— | 5.83/5.18 | 37.9/33.8 | —/1.0 | —/13.8 | 31/33 |
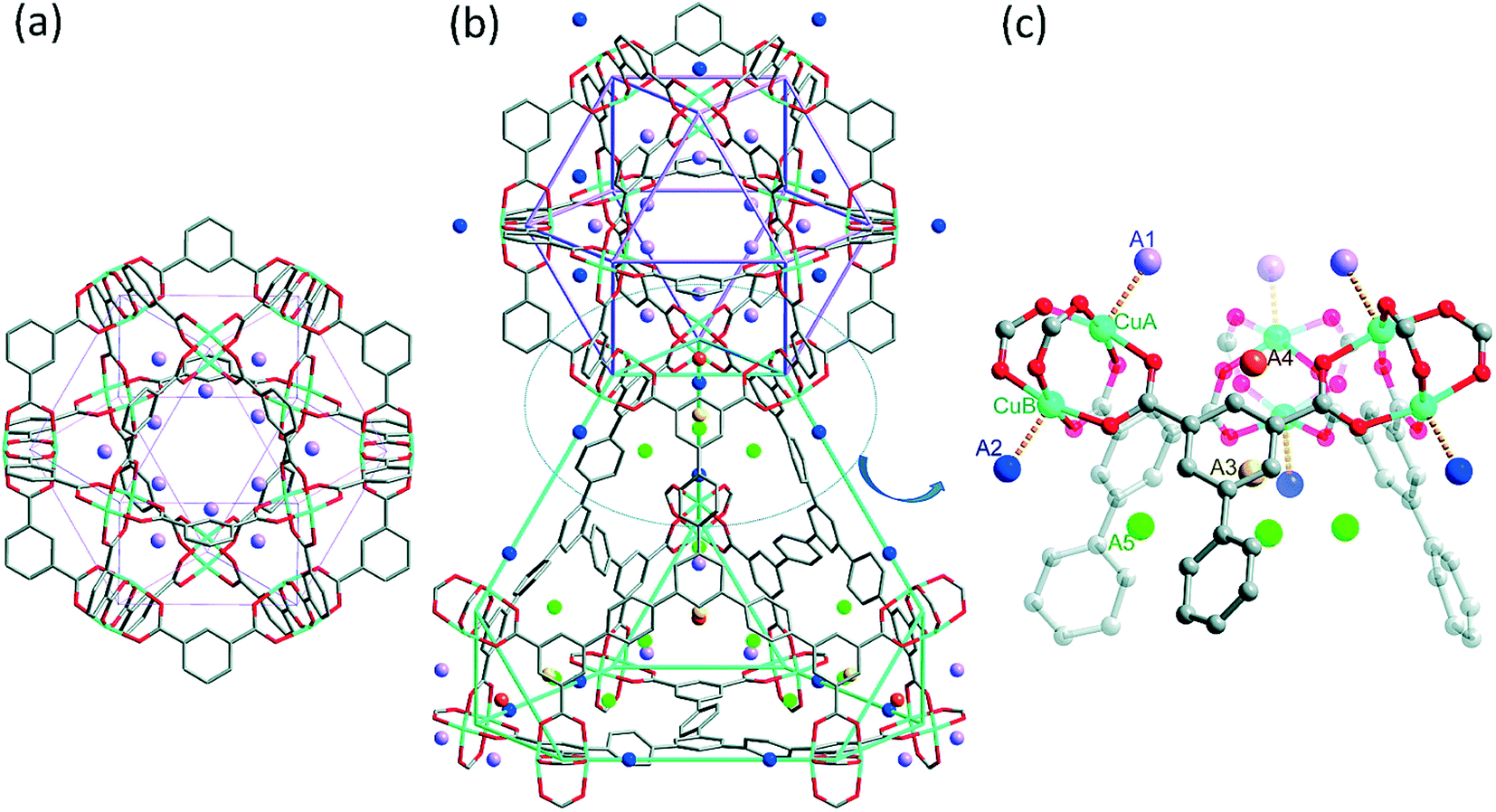 | ||
| Fig. 3 Views of the D2 molecule positions (colored spheres) in NOTT-112 as determined through neutron powder diffraction (NPD). (a) A view of the cub-Oh cage showing the D2 molecule positions at 0.5 D2/Cu loading. (b) A view of the cub-Oh and T-Td cages showing the D2 molecule positions at 2.0 D2/Cu loading. (c) A close-up view of the corner of the T-Td cage showing the five D2 molecule positions (A1, A2, A3, A4, and A5) at 2.0 D2/Cu loading. Atom colors: C = gray, O = red, Cu = turquoise, A1 = lavender, A2 = blue, A3 = yellow, A4 = orange, A5 = green. This figure was reproduced from ref. 13 within the guidelines provided by the American Chemical Society. Copyright 2010 American Chemical Society. | ||
Yan et al. discovered two different sorption sites in NOTT-112 at a loading of 0.5 D2/Cu (ca. 0.3 wt%).13 The first of these correspond to sorption onto the Cu2+ ions that are located within the cub-Oh cages in the MOF (site A1 in Fig. 3). The other sorption site was observed near the Cu2+ ions that are located within the T-Td and T-Oh cages (site A2 in Fig. 3). The authors denoted these Cu2+ ion sites as CuA and CuB, respectively. The refinements at this loading revealed that 85% of the D2 molecules sorbed onto the CuA ions. This suggests that both types of Cu2+ ions have different affinities toward the D2 molecule. This also indicates that the two Cu2+ ions that make up the copper paddlewheel units in NOTT-112 are located in unique environments in the MOF structure. Indeed, there are at least two chemically distinct M2+ ions in the crystal structure of all rht-MOFs. One type of M2+ ion faces toward the center of the linker and projects into the T-Td and T-Oh cages whereas the other type faces away from the center of the linker and projects into the cub-Oh cage. This is in contrast to HKUST-1 (ref. 42) and MOF-505 (ref. 43), two prototypical MOFs containing Cu2+ ions coordinated to 1,3,5-benzenetricarboxylate (BTC) and 3,3′,5,5′-biphenyltetracarboxylate (BPTC) linkers, respectively, as all metal ions in these MOFs are chemically equivalent.
The interaction distance between the center-of-mass (COM) of the sorbed D2 molecule and the CuA and CuB ions in NOTT-112 was found to be 2.23(1) and 2.41(1) Å, respectively, from the NPD studies.13 The shorter CuA–COM(D2) distance relative to that for the CuB ion indicates that the D2 molecules exhibit a stronger interaction about the CuA ions. Thus, the CuA ions represent the most favorable sorption sites in NOTT-112. We note that the CuB–COM(D2) distance is very similar to that for the corresponding interaction observed in HKUST-1 through NPD studies (2.39(1) Å).44
The authors hypothesized that the CuA ions are more favorable than the CuB ions in the MOF because the former types are located within the small cub-Oh cages.13 The finding that the CuA ions are preferred over the CuB ions at low loading has been supported through classical GCMC simulations of H2 sorption in NOTT-112 that were carried out by our group recently.45 It was discovered that the CuA ions exhibit a greater partial positive charge than the CuB ions through electronic structure calculations. This allowed for the H2 molecules to sorb initially onto the CuA ions in simulations involving explicit many-body polarization as the sorbate molecules were more attracted to the Cu2+ ion having the higher partial positive charge at low loading. Thus, the simulations suggest that the differing affinities that the two types of Cu2+ ions have toward sorbates are due to their distinct electrostatic profiles. Overall, the NPD studies of D2 sorption in NOTT-112 clearly demonstrated that the two Cu2+ ions that comprise the [Cu2(O2CR)4] clusters in rht-MOFs are chemically distinguishable and therefore results in unique binding sites about such MBBs.
At higher loadings, Yan et al. discovered additional D2 binding sites in NOTT-112 from their NPD experiments.13 These sorption sites are located in the corners of the T-Td cages in the MOF. At 1.0 D2/Cu loading, a third sorption site was identified between three isophthalate groups around the 3-fold axis of the triangular window connecting the cub-Oh and T-Td cages (site A3 in Fig. 3). A fourth sorption site was found in the region between three neighboring copper paddlewheel units at the same loading (site A4 in Fig. 3). This site is essentially located on the other side of the triangular window from the previous site, but on the same 3-fold axis. At a loading of 1.5 D2/Cu, a fifth sorption site was discovered, where the sorbate molecules localize near the phenyl rings of the linker in the T-Td cages (site A5 in Fig. 3). Note, these sorption sites within the corners of the T-Td cages were also observed through molecular simulation studies of H2 sorption in NOTT-112.45
B. Inelastic neutron scattering
INS is a valuable spectroscopic technique that is used to gain molecular level information on the H2 sorption sites in MOFs and other porous materials.46,47 Although NPD gives details on the crystal structure of the guest-filled material, INS provides information on the molecular excitations of H2 sorbed at different binding sites in the host. The INS spectra for H2 sorbed in a MOF contain a number of distinct peaks that arise from the rotational and translational excitations of the sorbate, where each transition typically corresponds to H2 sorbed at a specific binding site in the material. Note, most of the transitions observed in the INS spectra are due to the rotational excitations of the H2 molecule; this greatly simplifies the task of assigning peaks in the spectra. Tentative assignments of the peaks in the INS spectra can be made based on the observed neutron transfer energies, the presence of certain types and quantities of functionalities within the material, and trends within the H2 loading amount. Such assignments can be confirmed through quantum dynamics calculations on an accurate theoretical potential energy surface.47 To the best of our knowledge, only three rht-MOFs had INS studies performed on them to date: rht-MOF-1,25rht-MOF-4a,25 and rht-MOF-7.48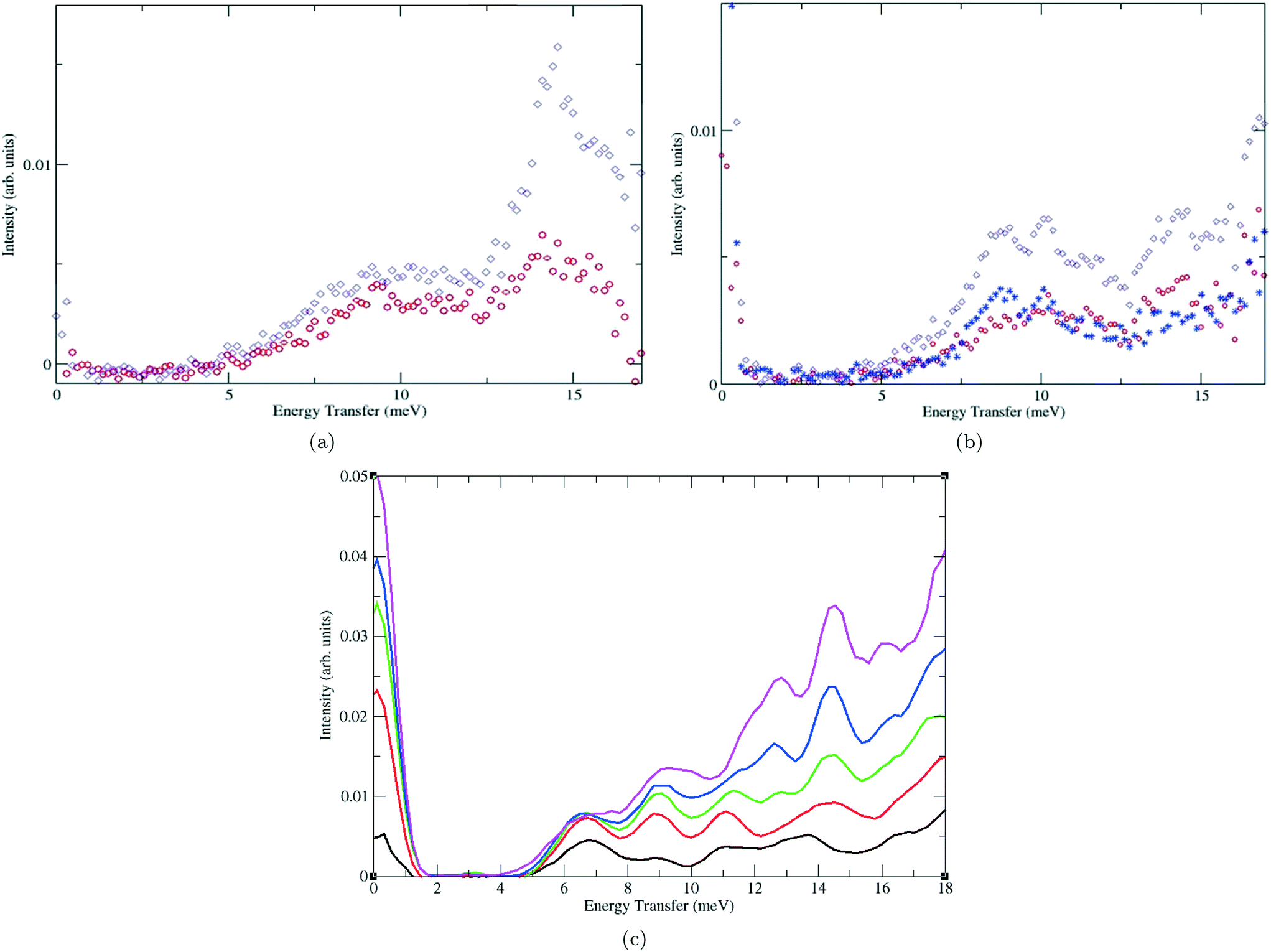 | ||
| Fig. 4 Inelastic neutron scattering (INS) spectra for H2 in (a) rht-MOF-1 at loadings of 1 H2/Cu (red) and 2 H2/Cu (violet), (b) rht-MOF-4a at loadings of 0.9 H2/Cu (red) and 1.5 H2/Cu (violet) with the difference spectrum shown in blue, and (c) rht-MOF-7 at loadings of 0.75 H2 per formula unit (black), 1.5 H2 per formula unit (red), 2.25 H2 per formula unit (green), 3 H2 per formula unit (blue), and 4 H2 per formula unit (magenta). The INS spectra for rht-MOF-1 and rht-MOF-4a were reproduced from ref. 25 within the guidelines provided by Wiley-VCH (Germany). Copyright 2012 Wiley-VCH (Germany). The INS spectra for rht-MOF-7 were reproduced from ref. 48 within the guidelines provided by the American Chemical Society. Copyright 2014 American Chemical Society. | ||
At both loadings, a peak can be observed at approximately 9.0 meV in the INS spectra. This peak most likely corresponds to H2 sorbing onto the Cu2+ ions of the copper paddlewheels on the basis of what was observed through INS measurements on other MOFs containing these MBBs.25,49–51 Indeed, INS studies on HKUST-1 also revealed a peak near 9.0 meV in the resulting spectra.50 Since both rht-MOF-1 and HKUST-1 contain [Cu2(O2CR)4] clusters, this peak at ca. 9.0 meV in the individual spectra can likely be attributed to H2 sorbing onto such sites. This was actually confirmed through two-dimensional quantum rotation calculations for a H2 molecule sorbed about the Cu2+ ion of the copper paddlewheels in both MOFs, as such calculations produced rotational levels that are near 9.0 meV for the lowest j = 0 to j = 1 transition.50,52 Note, in the absence of a barrier to rotation, a H2 molecule rotates freely with energy levels that are representative of a rigid rotor. The lowest j = 0 to j = 1 transition for unhindered H2 corresponds to a value of 14.7 meV.
At 2 H2/Cu loading, a well-defined shoulder ranging from 6.5–8.0 meV becomes apparent in the INS spectrum for rht-MOF-1. The authors in ref. 25 attributed this peak to H2 sorbing onto the Cu2+ ions of the [Cu3O(N4CR)3] units in the MOF. A peak within the aforementioned range of energies was not observed in the INS spectra for HKUST-1 since these Cu3O trimers are not present in this MOF.50 Thus, the assignment of the 6.5–8.0 meV peak to H2 sorbing onto the Cu2+ ions of the [Cu3O(N4CR)3] MBBs appears to be valid, especially since this was later confirmed through two-dimensional quantum rotation calculations.52 It appears that the Cu3O trimers become more occupied at higher loadings, which is consistent with what was observed through GCMC simulations of H2 sorption in this MOF.
The INS spectra for rht-MOF-1 also contain a small, but noticeable peak at approximately 5.0 meV for both loadings. This peak is associated with H2 sorbing onto the most favorable binding sites in the MOF. Note, in general, peaks that occur at lower neutron transfer energies in the INS spectra correspond to a higher barrier to rotation and therefore, a stronger interaction with the host. Two different GCMC simulations of H2 sorption in rht-MOF-1 revealed that the most preferential H2 sorption sites in the MOF are the NO3− counterions (see section III.A.1).52,53 Sorption onto these nitrate ions correspond to the initial H2Qst in the material as verified through simulations involving explicit many-body polarization.52
INS measurements on In-soc-MOF, another MOF possessing nitrate counterions,54 also contain a peak near 5.0 meV in the resulting spectra. The authors in that reference claimed that this peak should correspond to H2 sorbing onto the NO3− ions. Two-dimensional quantum rotation calculations for H2 sorbed onto the nitrate ions in both MOFs yielded a j = 0 to j = 1 transition of 5.63 and 5.78 meV for rht-MOF-1 (ref. 52) and In-soc-MOF (ref. 55), respectively. These calculations therefore suggest that the transition occurring at about 5.0 meV in the INS spectra for both MOFs is associated with sorption onto the nitrate counterions.
A large peak can be observed at about 14.0 meV in the INS spectrum for rht-MOF-1 at both loadings. This peak is associated with H2 sorbing onto the weaker binding sites in the MOF, most notably, the spacious interior of the cub-Oh cages according to GCMC simulations and attendant quantum dynamics calculations.52 The H2 molecules localized at such sites exhibit rigid rotor-like behavior since they give rise to transitions that are near the free rotor limit of 14.7 meV. As a result, these H2 molecules have very low barriers to rotation within the material.
As with the INS spectra for rht-MOF-1, a peak at about 9.0 meV can be observed in the spectrum for rht-MOF-4a at both loadings. This neutron transfer energy value is consistent with the lowest energy transition observed in other neutral MOFs containing copper paddlewheels.49–51 Thus, this peak at ca. 9 meV should correspond to H2 sorbing onto the Cu2+ ions of the copper paddlewheels in the MOF. The INS spectra for rht-MOF-4a also reveal a peak at around 10.5 meV. The authors in ref. 25 attributed this peak to sorption within the vicinity of the isophthalate moieties. A peak with high intensity was also observed at frequencies just below 13.5 meV, which could correspond to sorption near the alkoxy groups on the linkers. Note, unlike rht-MOF-1, quantum dynamics calculations were not performed on the H2 binding sites in rht-MOF-4a. Thus, the assignments of the peaks at approximately 10.5 and 13.5 meV in the INS spectra for this MOF are tentative and uncorroborated.
With regards to H2 sorption, Li et al. showed that rht-MOF-7 can sorb 2.65 wt% of H2 at 77 K/1 atm and has an initial Qst of 8.29 kJ mol−1.19 On the other hand, the corresponding values measured by Eddaoudi's group are 2.20 wt% and 6.77 kJ mol−1, respectively.48 INS measurements of H2 sorbed in rht-MOF-7 revealed a number of clearly defined peaks in the spectra, which are associated with different binding sites in the MOF. The INS spectra that were collected at various loadings for this MOF are shown in Fig. 4(c). The assignments of the peaks in this spectra have been confirmed through two-dimensional quantum rotation calculations on H2 sorbed at the binding sites that were discovered through GCMC simulations.
A relatively low energy peak can be observed in the INS spectra at around 6.8 meV. The low energy of this rotational tunnelling transition is indicative of a high barrier to rotation, and hence a very strong interaction with the host. Indeed, this peak is associated with H2 sorbed at the most favorable sorption site in rht-MOF-7. The intensity of this peak appears to saturate below a loading of 1.5 H2 per formula unit, which indicates that there is a limited number of these sites available in the MOF.
GCMC simulations of H2 sorption in rht-MOF-7 revealed that the most favorable binding site in the MOF is located between two neighboring Cu2+ ions that project into the T-Td and T-Oh cages,48 named the CuB ions according to the convention by Yan et al.13 An illustration of this site is shown in Fig. 5(a). The angular nature of the L7 linker causes two CuB ions to be in proximity to each other in the structure. Thus, as the H2 molecule is sorbed in this region, it can interact with the two adjacent CuB ions simultaneously, providing for a very favorable interaction. Two-dimensional quantum rotation calculations for H2 sorbed at this site produced a rotational level of 6.84 meV for the lowest transition, which is in excellent agreement with the lowest energy peak observed in the INS spectra for the MOF.48 Further, there are only 12 of these sites available within a unit cell of the MOF, which explains why the peak at ca. 6.8 meV is saturated below 1.5 H2 per formula unit loading.
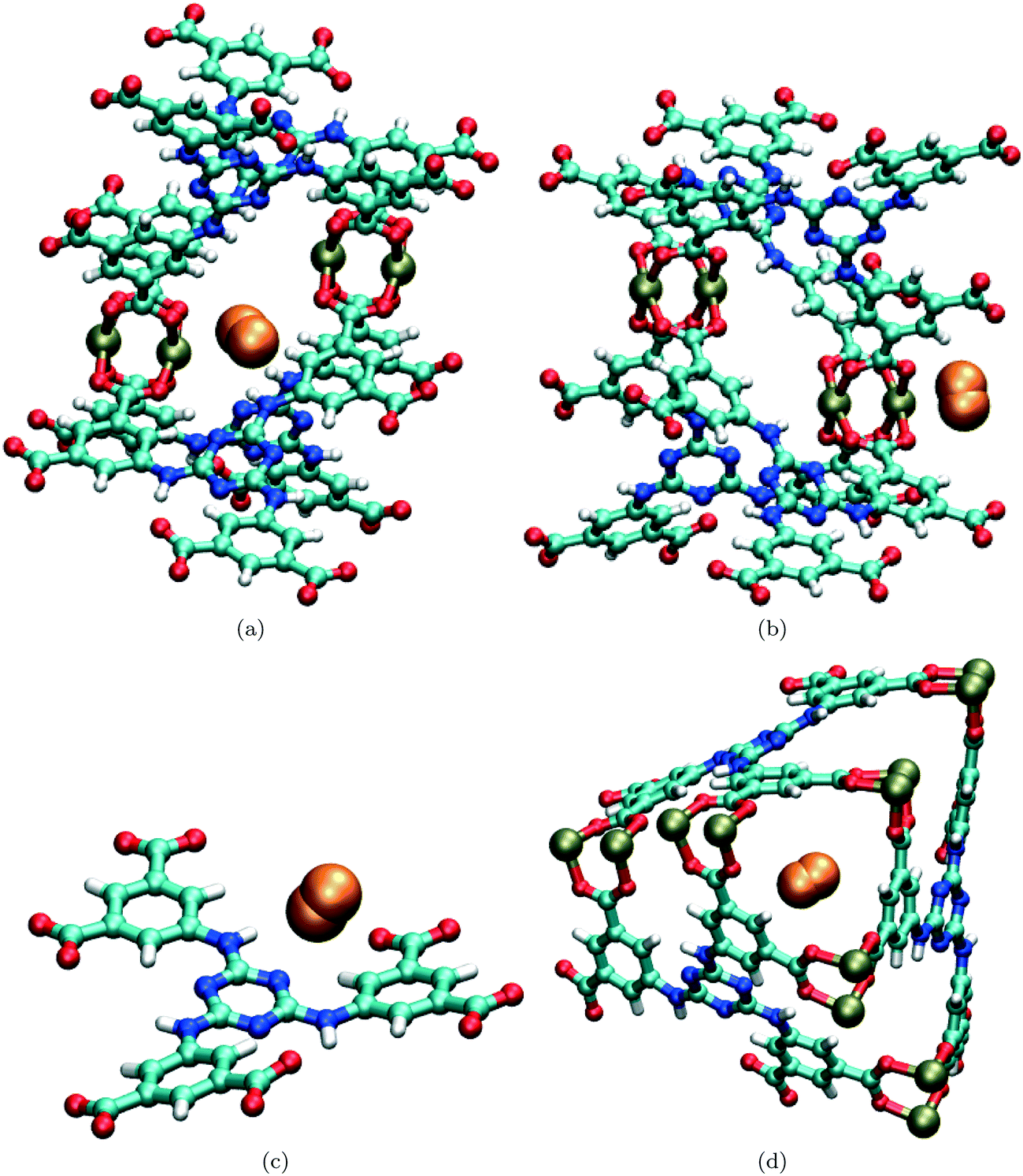 | ||
| Fig. 5 Molecular illustration of a H2 molecule (orange) sorbed at various sites in rht-MOF-7 as determined from the GCMC simulations performed in ref. 48: (a) between two CuB ions, (b) onto a single CuA ion, (c) onto the 1,3,5-triazine groups of the linkers, and (d) within the corner of the T-Td cage. These sorption sites correspond to the peaks observed at approximately 6.8, 9.0, 11.0 and 13.5 meV, respectively, in the INS spectra for the MOF (Fig. 4(c)). Atom colors: C = cyan, H = white, N = blue, O = red, Cu = tan. These figures were reproduced from ref. 48 within the guidelines provided by the American Chemical Society. Copyright 2014 American Chemical Society. | ||
The INS spectra show a peak at approximately 9.0 meV that gradually increases in intensity at higher loadings. This peak is typical for H2 sorbing onto a single Cu2+ ion of the copper paddlewheel unit as observed in rht-MOF-1 (Fig. 4(a)),25,52rht-MOF-4a (Fig. 4(b)),25 HKUST-1,50 and PCN-12.51 In the case of rht-MOF-7, the ca. 9.0 meV peak is associated with sorption onto the CuA ions, the Cu2+ ions that project into the cub-Oh cages in the MOF (Fig. 5(b)). This was indeed confirmed through two-dimensional quantum rotation calculations.48 Unlike the environment of the CuB ions, the CuA ions are much farther apart from each other. Thus, as the H2 molecule sorbs onto the CuA ions, it cannot interact with any other components of the MOF structure. This results in a relatively weaker interaction compared to sorption between two neighboring CuB ions. Note, the intensity of the 9.0 meV peak is nearly twice as high as that for the 6.8 meV peak at the highest loading measured (4 H2 per formula unit). This is consistent with the expected relative saturation amounts about both types of Cu2+ ions as there are two CuA sites for every region between two adjacent CuB ions.
Additional peaks can be observed at approximately 11.0 and 13.5 meV in the INS spectra for rht-MOF-7, especially at low loadings. GCMC simulations and two-dimensional quantum rotation calculations provided clarification on the identity of these sites.48 The center of the linker in the MOF can sorb up to three H2 molecules, with each sorbate interacting with a N atom on the 1,3,5-triazine group (Fig. 5(c)). This suggests that the peak at about 11.0 meV could correspond to sorption onto the 1,3,5-triazine moieties since the intensity of this peak at the highest loading is about three times as high as that for the lowest energy peak in the spectra. This was convincingly supported by two-dimensional quantum rotation calculations, as such calculations for H2 sorbed onto the 1,3,5-triazine groups yielded a j = 0 to j = 1 transition near 11 meV.
GCMC simulations of H2 sorption in rht-MOF-7 revealed that the H2 molecules also occupy the corners of the T-Td cages (Fig. 5(d)),48 similar to what was observed for D2 sorption in NOTT-112 through NPD (see section II.A).13 Two-dimensional quantum rotation calculations for H2 sorbed in this area generated rotational levels that are close to 13.5 meV for the two lowest j = 0 to j = 1 transitions.48 Thus, the peak at ca. 13.5 meV in the INS spectra should correspond to H2 sorbing within the corners of the T-Td cages. This also makes sense from a loading perspective since up to four H2 molecules can sorb within this region according to the GCMC simulations and the intensity of the 13.5 meV peak is approximately four times as high as that for the lowest energy peak.
Overall, INS studies of H2 sorbed in rht-MOF-1, rht-MOF-4a, and rht-MOF-7 revealed that the Cu2+ ions of the copper paddlewheels are the principal binding sites for H2 in these MOFs. This was exemplified by the fact that the spectra for all three MOFs contain a peak occurring at about 9.0 meV for all loadings measured. The presence of different functionalities in these rht-MOFs also gave rise to distinct features in the individual spectra. As demonstrated in the context of rht-MOF-1 (ref. 52) and rht-MOF-7 (ref. 48), the assignments of the peaks in the INS spectra can be verified through GCMC simulations and attendant quantum dynamics calculations.
III. Theoretical studies
A. Monte Carlo simulations
By far most of the elucidation of the binding sites in rht-MOFs have been made by way of Monte Carlo (MC) simulations, particularly within the grand canonical ensemble. Indeed, a number of GCMC simulation studies of gas sorption have been reported that yielded insights into the binding sites in these MOFs.27,28,33,45,48,52,56–61 We note that there are other GCMC simulation studies performed on rht-MOFs, particularly by Snurr and co-workers,14,23,24,29,30 but such works were more focused on showing the comparisons between experimental and theoretical observables.In short, GCMC simulations involve the random trial movement of sorbate molecules within a simulation box containing the MOF–sorbate system in which the chemical potential, volume, and temperature are kept fixed.62 Gibbs ensemble Monte Carlo (GEMC) is another technique that can be used to perform simulations of gas sorption in MOFs as demonstrated by Babarao et al.53 This method involves utilizing two simulation boxes, one for the MOF and another for the sorbates.63 The volume of the sorbent is kept fixed, but that for the sorbate is allowed to vary at a fixed pressure. In addition, the total number of sorbates in a GEMC simulation is kept fixed, but the sorbates can swap from one box to another. In this section, we discuss the sorption sites that were observed through selected MC simulation studies on various rht-MOFs.
As stated earlier (in section II.B.1), rht-MOF-1 is a cationic MOF that contains extra-framework nitrate counterions within the material. These NO3− ions are located within the cub-Oh cages in the solvated structure of the MOF. Molecular dynamics (MD) simulations revealed that these nitrate counterions migrate to the corners of the T-Td cages in the dehydrated structure; these regions therefore represent the equilibrium locations of the NO3− ions in the solvent-free material. The authors in ref. 53 carried out GEMC simulations on rht-MOF-1 with the nitrate ions positioned in the corners of the T-Td cages. Note, the aforementioned finding for the equilibrium locations of the NO3− ions in the dehydrated rht-MOF-1 was also reproduced from theoretical studies in our group.52
Babarao et al. evaluated the preferred H2 sorption sites in rht-MOF-1 by plotting the density contours of H2 sorbed in the MOF at 77 K and different pressures.53 The density contour plots that they generated at 77 K and 10, 100, and 1000 kPa are shown in Fig. 6. At 10 kPa, it can be observed that the H2 molecules mainly sorb onto the nitrate counterions that are located in the corners of the T-Td cages. This suggests that the unbound NO3− ions are the most favorable sorption sites for H2 in rht-MOF-1 since they are highly occupied at low loading.
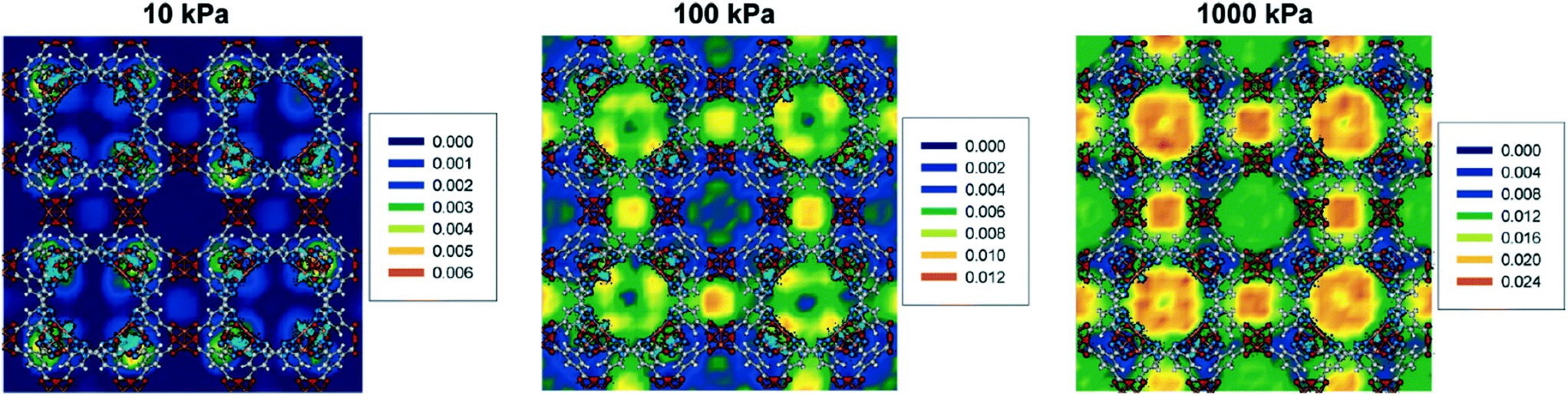 | ||
| Fig. 6 Density contours of H2 molecules in rht-MOF-1 at 77 K and 10 (left), 100 (middle), and 1000 kPa (right) based on the number of H2 molecules per Å3 according to the GEMC simulations performed in ref. 53. The COM distributions of NO3− counterions are depicted in cyan. Atom colors: C = gray, H = white, N = blue, O = red, Cu = peach. This figure was reproduced from ref. 53 within the guidelines provided by the American Chemical Society. Copyright 2010 American Chemical Society. | ||
At 100 kPa, the density contours reveal that the H2 molecules occupy the regions intersecting the cub-Oh and T-Oh cages as well as the cub-Oh and T-Td cages.53 The H2 molecules further populate these intersecting regions at 1000 kPa. Moreover, occupancy can be observed within the T-Td cages at this pressure. Interestingly, the COM distributions of NO3− ions are similar across all three pressures, implying that the positions of these counterions do not change significantly upon H2 sorption at 77 K and varying pressures.
Simulations of H2 sorption in rht-MOF-1 were also carried out by our group previously using GCMC methods.52 The three-dimensional histogram showing the most frequent sites of H2 occupancy in the MOF at 77 K and a very low pressure (0.001 atm) is displayed in Fig. 7. It can be observed that the majority of H2 molecules are sorbed onto the nitrate counterions within the corners of the T-Td cages at this loading. This is consistent with what was discovered by Babarao et al. from their simulations in rht-MOF-1 at low pressure.53 Indeed, these NO3− counterions represent the most favorable H2 sorption sites in the MOF since they are charged particles that increase the electrostatic field within the framework. It appears that they are more preferred than any of the open-metal sites in the MOF. Furthermore, sorption onto these counterions correspond to the initial H2Qst of the MOF as supported by our close reproduction of the experimental Qst plot from GCMC simulations.52
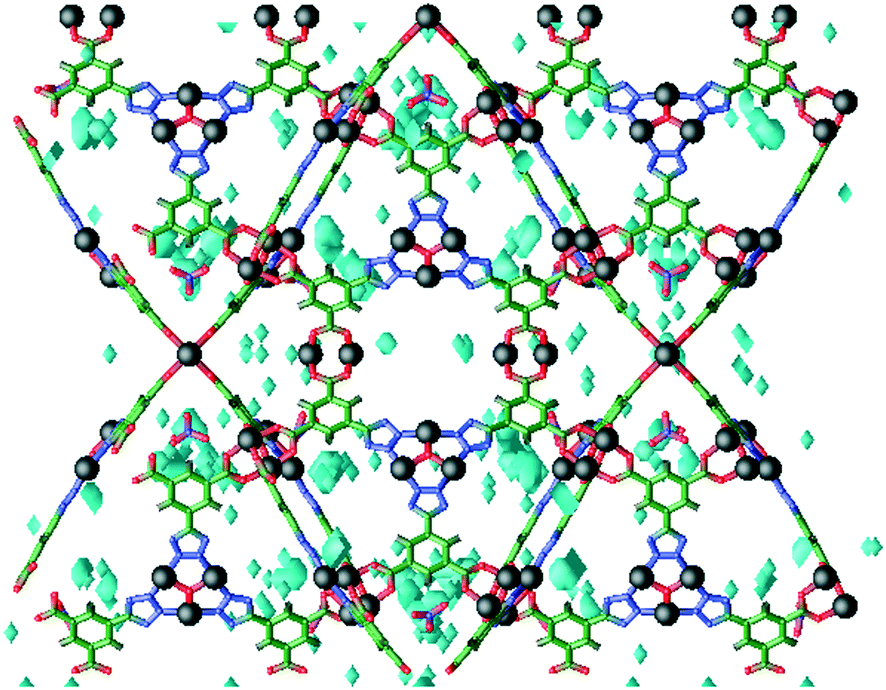 | ||
| Fig. 7 Three-dimensional histogram showing the most frequent sites of occupancy (cyan) for H2 molecules in rht-MOF-1 at 77 K and 0.001 atm according to the GCMC simulations performed in ref. 52. Atom colors: C = green, H = white, N = blue, O = red, Cu = black. This figure was reproduced from ref. 52 within the guidelines provided by the Royal Society of Chemistry. Copyright 2014 Royal Society of Chemistry. | ||
Note, in our theoretical study on rht-MOF-1, we also observed sorption onto the Cu2+ ions of the [Cu2(O2CR)4] and [Cu3O(N4CR)3] units.52 Such metal sites were captured in the simulations due to implementing many-body polarization interactions. After the NO3− counterions, the Cu2+ ions of the copper paddlewheels represent the next most favorable binding site, followed by the Cu2+ ions of the Cu3O trimers. This is consistent with what was discerned through INS studies on the MOF (see section II.B.1).25 These Cu2+ ion sites were not reproduced in the work of Babarao et al.,53 presumably due to the lack of explicit polarization effects in their simulations. At higher loadings, sorption was also identified within the cub-Oh cages in the MOF.52 The authors in ref. 53 also claimed to observe H2 sorbing into these cages at higher pressures.
Babarao et al. also examined the CO2 binding sites in rht-MOF-1 in their work.53 The authors stated that the CO2 sorption sites are similar to those shown in Fig. 6 for H2 sorption. However, they observed that the locations of the NO3− ions are more dispersed due to the greater thermal motion at 298 K. In addition to plotting the density contours and three-dimensional histograms and inspecting the modeled structure for binding sites, the preferential sorption sites in a MOF can be monitored through analysis of the radial distribution function (g(r)). Here, the normalized particle number for a sorbate is plotted at varying distances from a reference atom (usually from the host). This was demonstrated in ref. 53, where the authors evaluated the g(r) of CO2 about the nitrate ions in rht-MOF-1.
Fig. 8 shows the g(r) of CO2 carbon atoms about the N atoms of the NO3− ions in rht-MOF-1 at 298 K and different pressures (10, 100, and 1000 kPa). At 10 kPa, a significant peak can be observed at approximately 3.3 Å. This peak indicates a considerable amount of CO2 molecules sorbing onto the nitrate ions at low loading. Indeed, as in the case of H2, a very strong interaction exists between the CO2 molecules and the NO3− ions. Hence, these counterions represent favorable sorption sites for CO2 in rht-MOF-1. It can be observed that the magnitude of the peak at ca. 3.3 Å decreases at higher pressures. This is because, in addition to the nitrate ions, the CO2 molecules are sorbing onto other regions in the MOF structure at higher loadings, most notably, the regions intersecting the cub-Oh/T-Oh cages and cub-Oh/T-Td cages; this results in a lower relative occupancy about such counterions. Note, our group also used g(r) analysis to evaluate the favorable binding sites in rht-MOFs.36,45,48,52,56,58,59,61
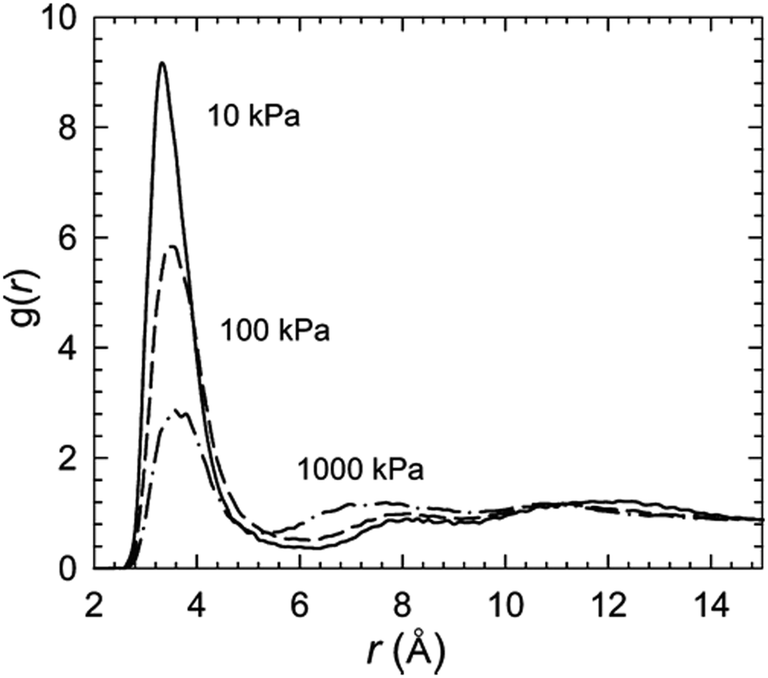 | ||
| Fig. 8 Radial distribution function (g(r)) of CO2 carbon atoms around the COM of the NO3− counterions in rht-MOF-1 at 298 K and 10 (solid), 100 (dashed), and 1000 kPa (solid and dotted) according to the GEMC simulations performed in ref. 53. This figure was reproduced from ref. 53 within the guidelines provided by the American Chemical Society. Copyright 2010 American Chemical Society. | ||
In our work on PCN-61,56 we showed that simulations using a polarizable H2 potential64 generated sorption isotherms and Qst values that are in outstanding agreement with the corresponding experimental measurements for the MOF. On the other hand, simulations utilizing potentials that neglect this interaction64,65 failed to reproduce experimental observables. Further, the inclusion of explicit many-body polarization effects was necessary to capture the sorption of H2 onto the open-metal sites in the MOF.56
Simulations involving explicit many-body polarization interactions allow for the binding sites to be discerned through the distribution of the induced dipoles on the sorbate molecules. This was first demonstrated in the MC simulation studies carried out by Belof et al. on In-soc-MOF.66 The normalized sorbate population is plotted as a function of the induced dipole magnitudes. The resulting dipole distribution for a sorbate in a MOF contain a number of different peaks, with each peak generally correlating to a region of occupancy in the material. In general, a peak that is found at high induced dipole magnitudes corresponds to sorption onto the most favorable binding sites in the MOF. This is because such sites induce high dipoles on the sorbate.
Fig. 9 shows the sorbate induced dipole magnitudes plotted against the normalized H2 population in PCN-61 at 77 K and various pressures (0.02, 0.15, 0.30, 0.50, and 1.0 atm) as obtained from simulations using a polarizable H2 potential in the MOF.56 A broad peak can be observed from about 0.25–0.50 D, which is most notable at the lowest loading considered (0.02 atm). This peak corresponds to H2 sorbing onto the exposed Cu2+ ions in the MOF as verified through examining the regions of sorbate occupancy for H2 molecules with these induced dipole magnitudes. Specifically, these H2 molecules were found to sorb onto the CuA ions as presented in Fig. 10(a). Indeed, the highly charged and polar Cu2+ ions of the copper paddlewheels induce rather high dipoles on the H2 molecules, which have a permanent dipole moment of 0 D in bulk on average.
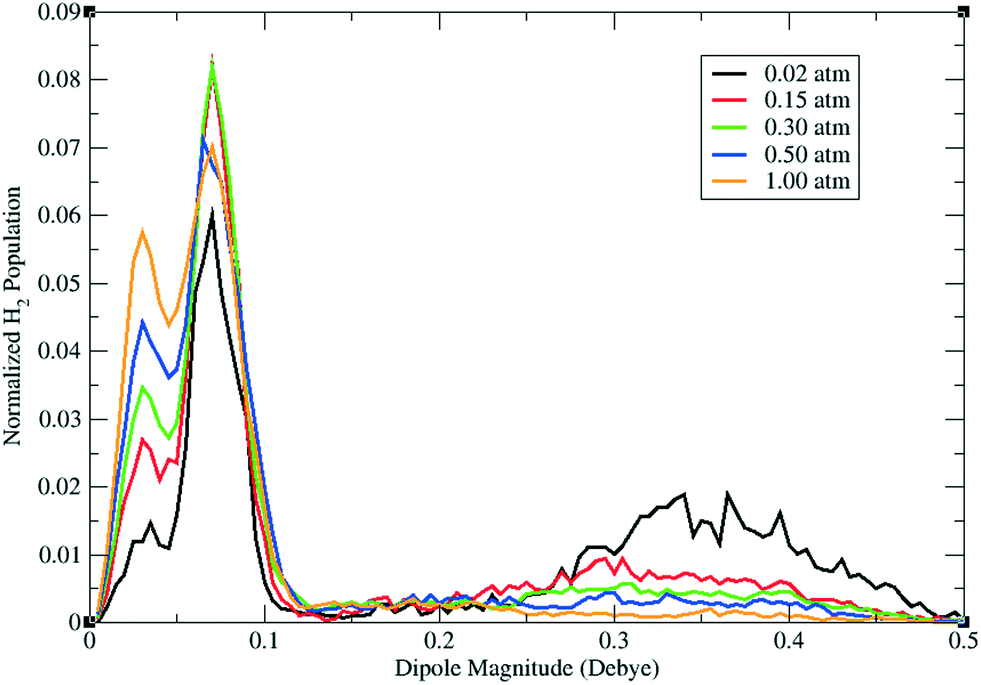 | ||
| Fig. 9 Normalized distribution of the induced dipoles on H2 molecules sorbed in PCN-61 at 77 K and various pressures (0.02 atm = black, 0.15 atm = red, 0.30 atm = green, 0.50 atm = blue, 1.00 atm = orange) according to GCMC simulations involving explicit many-body polarization performed in ref. 56. This figure was reproduced from ref. 56 within the guidelines provided by the American Chemical Society. Copyright 2012 American Chemical Society. | ||
 | ||
| Fig. 10 Three-dimensional histogram showing the initial H2 sorption sites (blue) in (a) PCN-61 and (b) Cu-TPBTM about the CuA and CuB ions, respectively, as determined from GCMC simulations involving explicit many-body polarization performed in ref. 56 and 58. Atom colors: C = green, H = white, N = blue, O = red, Cu = black. These figures were reproduced from ref. 58 within the guidelines provided by the American Chemical Society. Copyright 2013 American Chemical Society. | ||
A second peak can be observed from approximately 0.05–0.10 D in the H2 dipole distribution for PCN-61. Correlating this peak to regions of occupancy in the MOF revealed that H2 molecules with these induced dipoles sorb into the corners of the T-Td cages.56 This area was also discovered as a sorption site in other neutral rht-MOFs, such as NOTT-112 (section II.A)13 and rht-MOF-7 (section II.B.3).48 Note, such induced dipole magnitudes are comparable to that for the permanent dipole on CO (0.122 D),67 which has a normal boiling point of 82 K. An additional peak that increases in intensity at higher pressures can be observed from 0.0–0.05 D. This peak corresponds to H2 molecules crowding into the more spacious regions of the MOF (e.g., the center of the T-Td and T-Oh cages).56 As H2 molecules are sorbed into such areas, the sorbates exhibit lower induced dipoles since they are nearly representative of “free” H2.
Although Cu-TPBTM and PCN-61 are highly isostructural, differing only in the functionality located between the isophthalate groups and the central aromatic ring of the linker (as well as minor differences in the unit cell lengths, pore sizes, etc.), they exhibit distinct gas sorption mechanisms at low loading according to the GCMC simulations performed by our group.58 As with PCN-61, we showed that implementing classical polarization was requisite for reproducing experimental observables and capturing the sorption of H2 onto the open-metal sites in Cu-TPBTM. However, ab initio calculations revealed that Cu-TPBTM exhibits a different charge distribution about the copper paddlewheels compared to PCN-61; this had an effect on the initial binding sites in both MOFs.
Electronic structure calculations on different representational fragments of PCN-61 indicate that the CuA ions, the Cu2+ ions projecting into the cub-Oh cages in the MOF, display a greater partial positive charge relative to the CuB ions.56 Analogous calculations on Cu-TPBTM revealed the opposite trend, where it was discovered that the CuB ions, the Cu2+ ions projecting into the T-Td and T-Oh cages, exhibit the higher partial positive charge.58 The CuA ions have a greater partial positive charge in PCN-61 probably due to the fact that they are located in a more favorable chemical environment in general by projecting into the cub-Oh cages. However, in Cu-TPBTM, it appears that the presence of the negatively charged amide O atom on the linker causes the partial positive charge of the CuB ions to increase through an inductive effect (and thereby decreasing its electron density) since the amide groups are proximal to such Cu2+ ions in the structure. This could explain why the partial positive charge is greater on the CuB ions compared to the CuA ions in this MOF.
As explained briefly for NOTT-112 in section II.A., the sorbate molecules are more attracted to the type of Cu2+ ion having the higher partial positive charge in rht-MOFs. Indeed, GCMC simulations of H2 sorption in PCN-61 and Cu-TPBTM with the inclusion of explicit many-body polarization interactions revealed that the H2 molecules sorbed initially onto the CuA ions in the former (Fig. 10(a)),56 and onto the CuB ions in the latter (Fig. 10(b)).58 In other words, for the primary sorption site, the H2 molecules bind onto the Cu2+ ions located inside the cub-Oh cages in PCN-61, whereas they sorb onto the Cu2+ ions located outside the cub-Oh cages in Cu-TPBTM. We note that sorption of H2 was also observed within the corners of the T-Td cages and onto the amide groups in Cu-TPBTM according to GCMC simulations.58 Additionally, a similar phenomenon was observed in the case of CO2 sorption in both of these rht-MOFs.61
 | ||
| Fig. 11 Density distribution profiles between CO2 and the COM of rht-MOF-7 at 298 K and 10 (black) and 100 kPa (red) according to the GCMC simulations performed in ref. 57. This figure was reproduced from ref. 57 within the guidelines provided by the American Chemical Society. Copyright 2012 American Chemical Society. | ||
GCMC simulations of gas sorption were also carried out in rht-MOF-7 by our group in 2014, where the CO2 and H2 sorption mechanism and binding sites in the MOF were investigated.48 It was shown that the simulated CO2 and H2 sorption isotherms and Qst values were in close agreement with the experimental measurements reported by Eddaoudi and co-workers.20 These simulations were executed using polarizable potentials for the respective sorbates.64,69 The inclusion of explicit many-body polarization interactions in simulation allowed for a significant quantity of CO2 and H2 molecules to sorb onto the open-metal sites in the MOF at low loading.48
A number of distinct CO2 binding sites were observed in rht-MOF-7 from the simulations performed by our group.48 The most favorable of these corresponds to the sorption of CO2 between two neighboring CuB ions, where each oxygen atom of the sorbate interacts with a single Cu2+ ion of adjacent copper paddlewheels (Fig. 12(a)). This sorption site is actually an example of a single-molecule trap for CO2, which has been similarly observed in other MOFs.70,71 The small and angular nature of the L7 linkers permit two neighboring [Cu2(O2CR)4] clusters to be in proximity to each other, thus allowing this sorption site to exist in the material. Further, this binding site corresponds to the initial CO2Qst value for the MOF (ca. 45 kJ mol−1)20 as validated through our classical GCMC simulations involving explicit polarization.48 Indeed, as demonstrated in the context of rht-MOF-7, the current smallest member of the rht-MOF family, tuning the pore sizes in MOFs can lead to optimal interactions between the sorbate molecules and the framework.
 | ||
| Fig. 12 Molecular illustration of a CO2 molecule sorbed (a) between two CuB ions and (b) onto a single CuA ion in rht-MOF-7 as determined from the GCMC simulations performed in ref. 48. Atom colors: C = cyan, H = white, N = blue, O = red, Cu = tan. These figures were reproduced from ref. 48 within the guidelines provided by the American Chemical Society. Copyright 2014 American Chemical Society. | ||
The next most preferrential sorption sites for CO2 in rht-MOF-7 are the CuA ions.48 An illustration of a CO2 molecule sorbing onto this site is shown in Fig. 12(b). Unlike what was observed about the CuB ions, a CO2 molecule can only sorb onto a single CuA ion since such Cu2+ ions are positioned farther apart from each other as a result of being located in the cub-Oh cages. The interaction for sorption onto a CuA ion is weaker than that for binding between two adjacent CuB ions. GCMC simulations revealed that the corners of the T-Td and T-Oh cages and the 1,3,5-triazine groups are also sites of occupancy for the CO2 molecules in the MOF, especially at higher loadings. These CO2 sorption sites were also discovered in the simulation studies performed by Jiang's group in ref. 60, which was the third paper that reported theoretical gas sorption results in rht-MOF-7. Note, the preferred H2 binding sites in the MOF were also identified from our simulations;48 these sites are discussed in section II.B.3 and shown in Fig. 5.
The measured CO2 uptakes for the MOF by the three groups at 298 K/1 atm are similar to each other (ca. 4.2, 4.6, and 4.0 mmol g−1 in ref. 27–29, respectively). When comparing to other MOFs within the rht-MOF platform, only rht-MOF-7,19rht-MOF-9,31,33 and Cu-TPBTM15 have higher experimental CO2 uptakes under the same conditions to the best of our knowledge. In addition, according to the empirical fitting by Wang et al.,27 NTU-105 exhibits an initial CO2Qst of about 36 kJ mol−1, which is third to only rht-MOF-7 (ref. 19 and 20) and rht-MOF-9 (ref. 31) in the rht-MOF family. The same authors demonstrated that the MOF has a H2 uptake of 2.75 wt% at 77 K/1 atm and a zero-loading Qst value of roughly 6.6 kJ mol−1.27 This value for the H2 uptake is among the highest of reported rht-MOFs at the same state point.27,34 NTU-105 was also shown to be one of the top sorbents for CH4 storage.29,72
The three experimental references for NTU-105 also list the results from GCMC simulations of gas sorption in the MOF.27–29 All three groups reported simulated gas sorption isotherms in decent agreement with their experimental measurements using nonpolarizable potentials.68,73 Attempts at elucidating the favorable binding sites for CO2 in NTU-105 were made by Wang et al.27 and Yan et al.28 The former investigated the interactions between the CO2 molecules and the MOF by analyzing the g(r) of these sorbates around different framework atoms.27Fig. 13 shows the g(r) of CO2 carbon atoms about the Cu2+ ions of the copper paddlewheels and the various N atoms of the 1,2,3-triazole groups of the linker in NTU-105 at 298 K and three different pressures (1, 10, and 100 kPa). The three distinct N atoms are denoted N1, N2, and N3, where N1 represents the N atom connected to the isophthalate group and N3 is the N atom that is closest to the central aromatic ring. Note, unlike what was implemented in other GCMC simulation studies on rht-MOFs,28,33,45,48,52,56,58–61 the simulations in ref. 27 treated all Cu2+ ions as chemically equivalent.
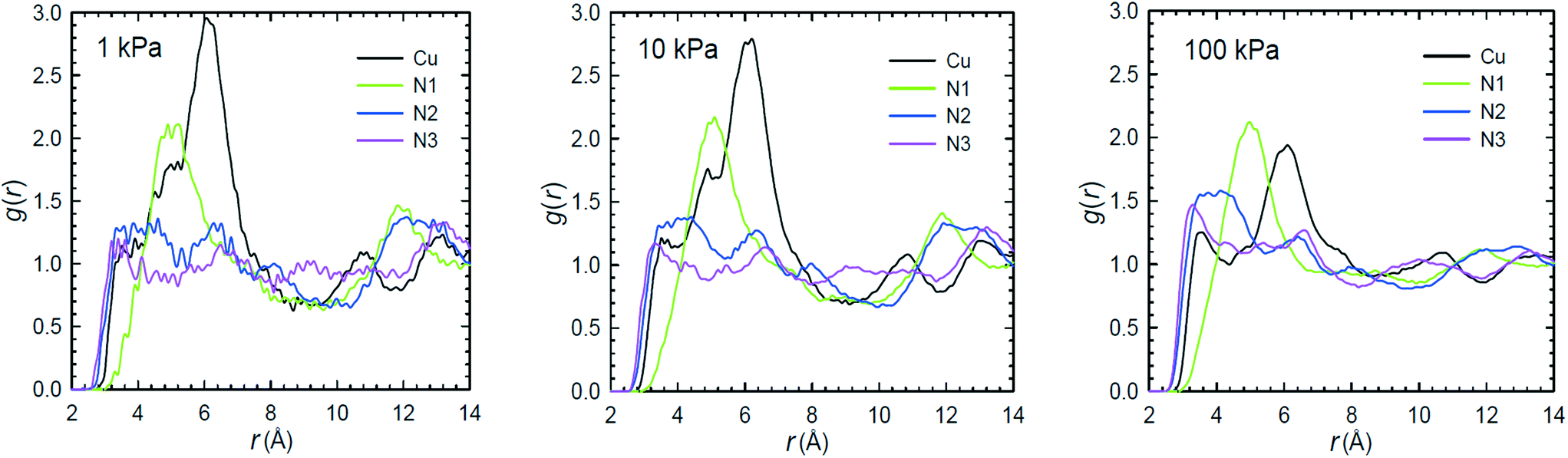 | ||
| Fig. 13 Radial distribution function (g(r)) of CO2 carbon atoms around the Cu2+ ions (black) and various N atoms on the 1,2,3-triazole group of the linker (N1 = green, N2 = blue, N3 = magenta) in NTU-105 at 298 K and 1 (left), 10 (middle), and 100 kPa (right) according to the GCMC simulations performed in ref. 27. This figure was reproduced from ref. 27 within the guidelines provided by the Nature Publishing Group. Copyright 2013 Nature Publishing Group. | ||
The g(r) of CO2 carbon atoms about the Cu2+ ions at all three pressures reveal a modest nearest-neighbor peak ranging from ca. 3.2–3.6 Å. This suggests that there are some CO2 molecules sorbing onto the open-metal sites in NTU-105 within the simulations. The aforementioned range for the Cu2+–C(CO2) interaction is comparable to that observed in HKUST-1 through NPD studies.74 The significant peak spanning from approximately 5.2–6.8 Å (maximum at 6.0 Å) in the g(r) around the metals probably corresponds to CO2 localizing in the corners of the T-Td cages. Additionally, it can be deduced that the CO2 molecules display strong affinity toward the N1 atoms as exemplified by the large peak ranging from ca. 4.4–6.0 Å in the g(r) about such atoms across all pressures. The CO2 molecules also exhibit favorability toward the N2 and N3 atoms since the g(r) of sorbates about these N atoms reveal a nearest-neighbor peak occurring at short distances.
Yan et al. discovered a number of different CO2 sorption sites in NTU-105 from their GCMC simulations by inspecting the modeled structure at 298 K and various pressures.28 At 10 kPa, the authors identified that the CO2 molecules initially sorb onto the CuA ions in their simulations, which they denoted site I (Fig. 14(a)). This site is analogous to what is depicted in Fig. 12(b) for rht-MOF-7. At the same pressure, two additional CO2 sorption sites were observed, which the authors referred to as sites II and III (Fig. 14(b)). Site II corresponds to the sorption of CO2 in the center of the triangular-shaped window formed by three isophthalate moieties and three [Cu2(O2CR)4] clusters in the T-Td cage. This site is essentially located in the corner of the T-Td cage, which has been observed as a favorable binding site for the sorbate in other rht-MOFs through MC simulations.48,53,59,61 Site III is located within the region formed by the peripheral arms from three different L8 linkers in the T-Td cage, where the CO2 is proximal to the N1 atom. This site is consistent with what was discerned in the g(r) of CO2 molecules about the N1 atoms in the GCMC simulation studies executed by Wang et al.27
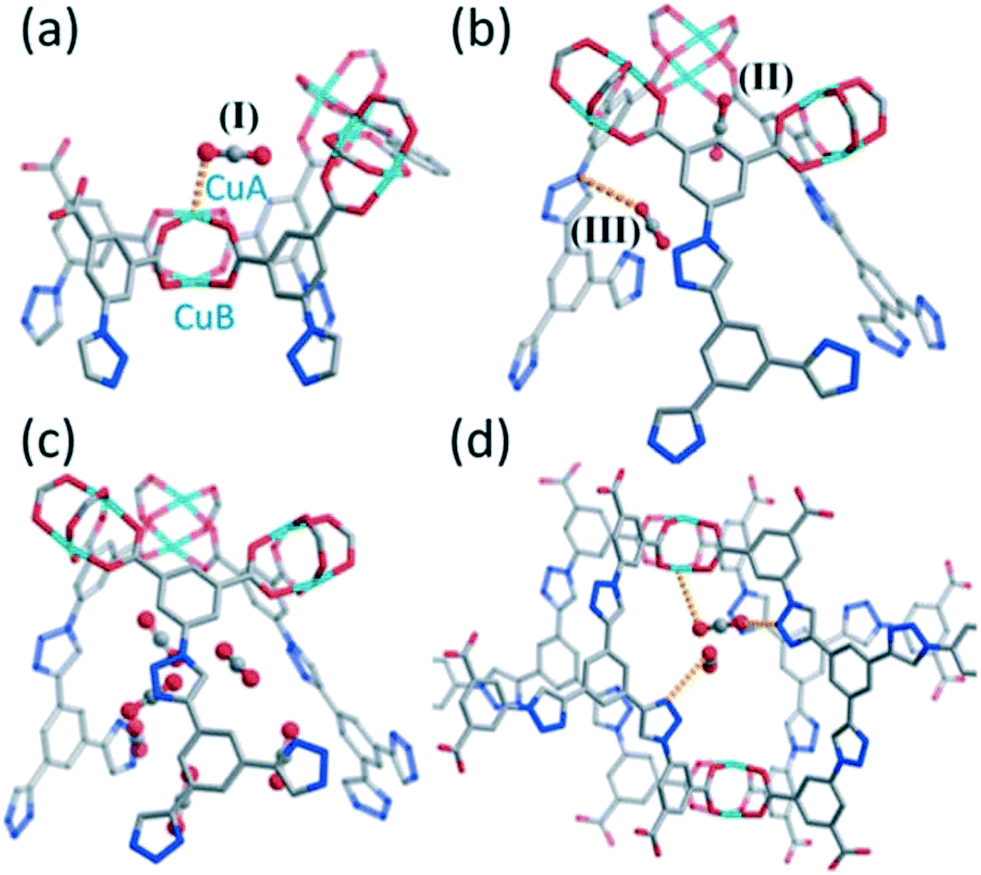 | ||
| Fig. 14 Molecular illustration of the CO2 sorption sites in NTU-105 at 298 K and ((a) and (b)) 10 kPa and ((c) and (d)) 50 kPa as determined from the GCMC simulations performed in ref. 28: (a) onto a single CuA ion (site I), (b) at sites II and III (see text in section III.A.5), (c) within the T-Td cages, and (d) within the small cavity created by four L8 linkers connecting the T-Td and T-Oh cages. Atom colors: C = gray, N = blue, O = red, Cu = cyan. This figure was reproduced from ref. 28 within the guidelines provided by the Royal Society of Chemistry. Copyright 2013 Royal Society of Chemistry. | ||
At a higher pressure (50 kPa), the authors in ref. 28 observed that the CO2 molecules crowd into the spacious region of the T-Td cage (Fig. 14(c)) and the small cavity surrounded by four L8 linkers connecting the T-Td and T-Oh cages (Fig. 14(d)). They also identified a significant amount of CO2 molecules sorbing in the cub-Oh cages at this pressure. Further, at pressures of 50 and 100 kPa, their simulations revealed a chain of CO2 molecules forming through the Cu2+–CO2, 1,2,3-triazole–CO2, and CO2–CO2 interactions in the MOF (Fig. 15). Overall, the results from GCMC simulations performed by Wang et al.27 and Yan et al.28 confirmed that the presence of open-metal sites and polar 1,2,3-triazole groups in NTU-105 contributes to high CO2 uptake in the material.
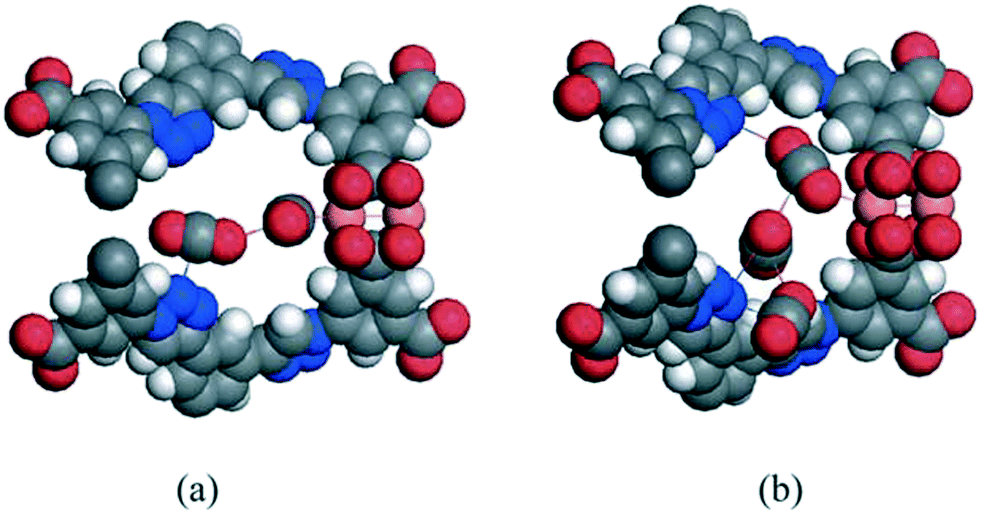 | ||
| Fig. 15 Molecular illustration of the preferrential CO2 sorption sites in NTU-105 at 298 K and (a) 50 kPa and (b) 100 kPa as determined from the GCMC simulations performed in ref. 28. Atom colors: C = gray, H = white, N = blue, O = red, Cu = peach. This figure was reproduced from ref. 28 within the guidelines provided by the Royal Society of Chemistry. Copyright 2013 Royal Society of Chemistry. | ||
B. Electronic structure calculations
In contrast to MC simulations, very few studies utilized electronic structure methods (e.g., ab initio and density functional theory (DFT)) to investigate the binding sites in rht-MOFs. This is likely because of the computational expense involved with implementing such quantum mechanical calculations on these MOFs. For example, periodic DFT calculations for a sorbate molecule localized in an rht-MOF are in fact computationally prohibitive due to the very large size of the unit cells of these materials. Thus, it is much more feasible to determine the sorption sites in these MOFs through classical MC simulations. However, electronic structure calculations can be used to assess the binding strength for a guest molecule sorbing about certain components or functionalities within the rht-MOF structure. These calculations typically involve optimizing the position of a sorbate molecule localized about a properly truncated cluster taken from the unit cell of the MOF and subsequently evaluating the binding energy. The magnitude of the calculated binding energy can provide indication on whether the sorption site is favorable in the MOF. To the best of our knowledge, we only know of electronic structure calculations of the sorbent–sorbate interaction performed in two studies for rht-MOFs: rht-MOF-9 in ref. 33 and rht-MOF-1 and rht-MOF-pyr in ref. 36.The authors in ref. 33 carried out GCMC simulations and DFT calculations in order to gain information on the CO2 sorption sites in rht-MOF-9. Their GCMC simulations revealed that the CO2 molecules prefer to sorb onto the Cu2+ ions of the copper paddlewheels and the secondary amine groups on the linker. In order to examine the role of the s-heptazine groups toward CO2 sorption, Liu et al. carried out DFT calculations of a CO2 molecule interacting with the secondary amine group on a truncated organic linker of the MOF and two other hypothetical variants of the linker in which the N content of the s-heptazine ring decreased sequentially. The calculations were performed at the 6-311++G(d,p)/B3LYP level of theory using the Gaussian09 package.75 The optimized distances between the CO2 oxygen atom and H atom of the secondary amine group as well as the calculated binding energies for the CO2–HNR2 interaction for all three model compounds are shown in Fig. 16.
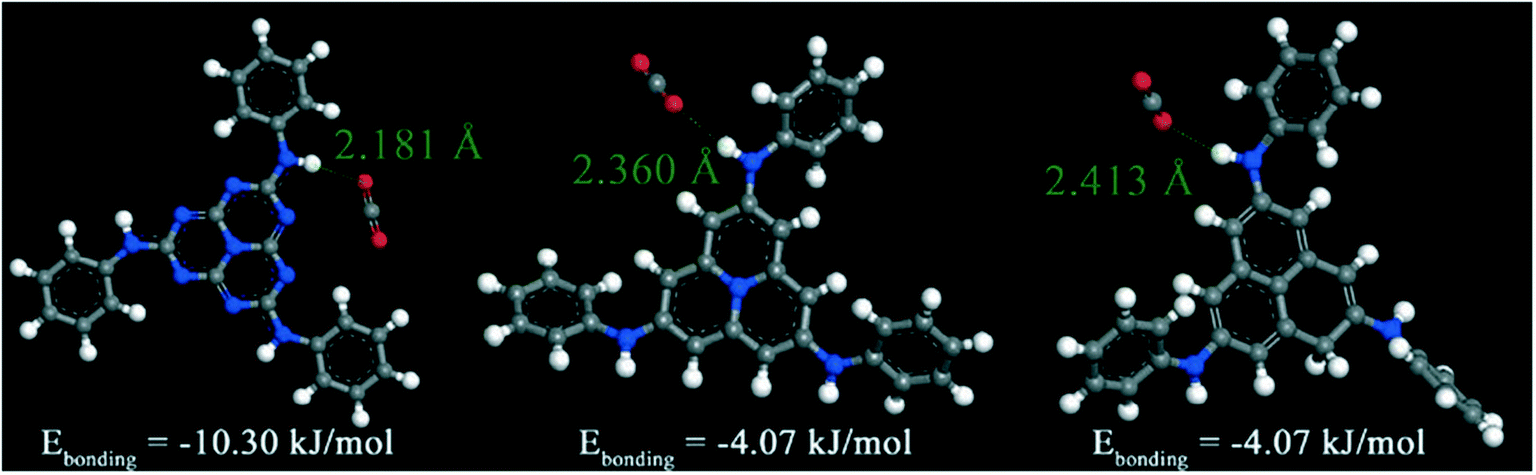 | ||
| Fig. 16 Optimized position of a CO2 molecule about the secondary amine groups within the truncated linker in rht-MOF-9 as well as different analogues of the s-heptazine unit as determined from the DFT calculations performed in ref. 33. The distances between the CO2 oxygen atom and the H atom of the secondary amine group are shown along with the calculated binding energies. Atom colors: C = gray, H = white, N = blue, O = red. This figure was reproduced from ref. 33 within the guidelines provided by the Royal Society of Chemistry. Copyright 2014 Royal Society of Chemistry. | ||
Geometry optimizations revealed that the interaction distance between the CO2 oxygen atom and the secondary amine H atom decreases with increasing number of N atoms on the polycylic aromatic ring. The binding energy associated with the CO2–HNR2 interaction for the truncated linker of rht-MOF-9 was calculated to be −10.3 kJ mol−1, while those for the other two linkers are notably lower (both at −4.07 kJ mol−1), indicating weaker guest–host interactions.33 It is clear from these comparisons that the electronegative O atoms of the CO2 molecule prefer to bind to the electropositive H atoms of the secondary amine group on the linker. However, the authors in ref. 33 attributed the stronger CO2 affinity for the truncated linker of rht-MOF-9 to the greater density of N atoms on the polycylic aromatic ring, namely a synergistic effect with the N atoms that are proximal to the CO2–HNR2 interaction. Indeed, as the CO2 molecule sorbs onto a secondary amine group of the L9 linker in the MOF, the positively charged C atom of the sorbate can also interact with the negatively charged N atom of the s-heptazine group that is nearby. We note that the richness of N atoms in rht-MOFs does not always lead to an increase in CO2 sorption affinity as explained in the next section.
Despite being highly isostructural to rht-MOF-1, rht-MOF-pyr was shown to display a much greater stability in moisture, water, steam, and acid.35 Indeed, this MOF retained its crystallinity after being exposed to ambient air with a relative humidity (RH) of ca. 70% for a week, water for 15 days at room temperature, steam (100% RH at 100 °C) for 6 hours, and aqueous HCl solution with a pH of 2.5 for two weeks at 298 K. The BET surface area of rht-MOF-pyr did not change significantly after being subjected to the aforementioned conditions. Many other rht-MOFs do not display the same type of stability as they lose their crystallinity after prolonged exposure to air.31,35 According to the electronic structure calculations performed by our group, the improved stability for rht-MOF-pyr relative to rht-MOF-1 can be attributed to the increase in electron density of the N atoms that are coordinated to the Cu2+ ions of the Cu3O trimers, which resulted in stronger bonds with such Cu2+ ions.35 This in turn lead to an increase in the stability about the [Cu2(O2CR)4] clusters. Note, an analogue containing 1,2,3-triazole groups was also synthesized in ref. 35 and shown to exhibit water and chemical stability.
rht-MOF-pyr was shown to display higher atmospheric CO2 uptake than rht-MOF-1 at 273 and 298 K according to experimental measurements.36 The former also exhibited a greater CO2/CH4 selectivity at 298 K/1 atm on the basis of ideal adsorbed solution theory (IAST) calculations.76 These results seem counterintuitive as it was expected that rht-MOF-1 would display higher CO2 uptake and selectivity due to possessing more N atoms on the framework. However, as demonstrated from GCMC simulations and DFT calculations carried out by our group, the local electric field of the [Cu3O(N4−x(CH)xCR)3] (x = 0 or 2) units in the two MOFs played a more dominant role toward CO2 sorption than the number of exposed N atoms on the linkers.36
GCMC simulations of CO2 sorption in rht-MOF-1 and rht-MOF-pyr identified the NO3− counterions, the Cu2+ ions of the copper paddlewheels, and the Cu2+ ions of the Cu3O trimers as favorable binding sites for the sorbate in both MOFs.36 This is consistent with what was observed for H2 sorption in rht-MOF-1 through related studies.52 Since the main difference in the CO2 uptakes among the two MOFs was expected to be due to the interaction between the CO2 molecules and the Cu3O trimers, additional calculations about such units were implemented, one of which included DFT calculations for a single CO2 molecule positioned about the [Cu3O(N4−x(CH)xCR)3] (x = 0 or 2) clusters for both rht-MOFs.36 Such electronic structure calculations were performed to investigate the significance of substituting tetrazole with pyrazole on CO2 sorption and to assess the difference in the binding energies about these MBBs.
The DFT calculations executed in ref. 36 utilized a truncated hexatopic building unit for both rht-MOFs, where all carboxylate groups of the L1 and L2 linkers were replaced with optimized H atoms. The CO2 molecule was optimized to an energetically favorable position within both model compounds using the 6-31G* basis set for all C, H, N, and O atoms, the LANL2DZ effective core potential basis set77 for the Cu2+ ions, and the B3LYP functional.78,79 These calculations were performed with the NWChem ab initio software.80 The optimized positions for a CO2 molecule about a Cu2+ ion of the truncated hexatopic building unit for rht-MOF-1 and rht-MOF-pyr are shown in Fig. 17. The calculations revealed nearly equivalent distances for the Cu2+–O(CO2) interaction, but notable differences in the orientation of the CO2 molecule about these units.
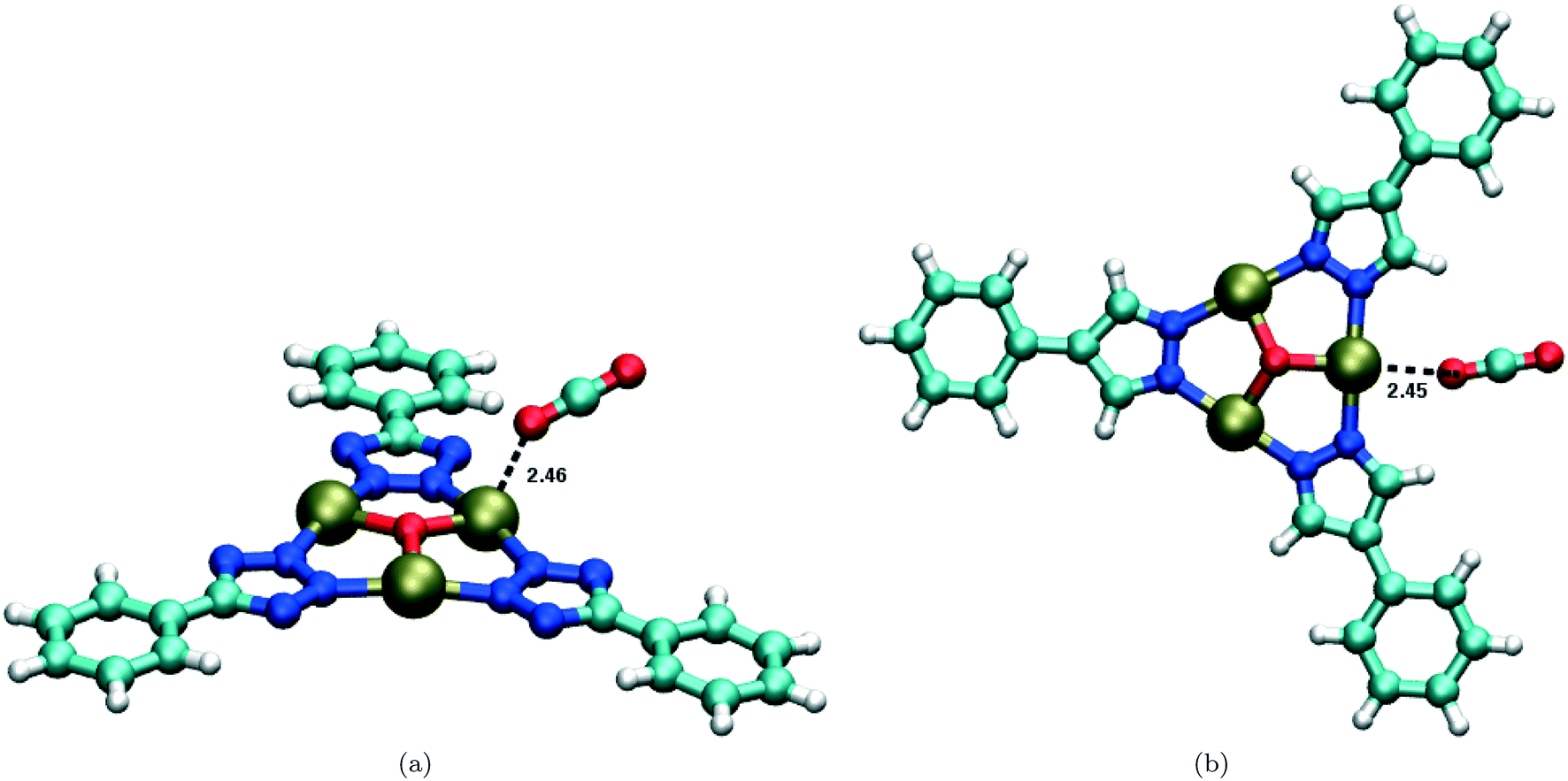 | ||
| Fig. 17 Optimized position of a CO2 molecule about the Cu3O trimer within the truncated hexatopic building unit in (a) rht-MOF-1 and (b) rht-MOF-pyr as determined from the DFT calculations performed in ref. 36. The distances (in Å) between the Cu2+ ion and the closest O atom of the CO2 molecule are also shown. Atom colors: C = cyan, H = white, N = blue, O = red, Cu = tan. These figures were reproduced from ref. 36 within the guidelines provided by the Royal Society of Chemistry. Copyright 2015 Royal Society of Chemistry. | ||
It was observed that as the oxygen atom of the CO2 molecule binds onto the Cu2+ ion of the Cu3O trimer in rht-MOF-pyr, it can also interact with the nearby positively charged H atoms of the pyrazole groups simultaneously (Fig. 17(b)).36 This synchronized binding keeps the sorbate molecule in-plane with respect to the [Cu3O(N2(CH)2CR)3] trimers, resulting in a favorable interaction. In contrast, as the CO2 oxygen atom sorbs onto the Cu2+ ion of the trimer in rht-MOF-1, it is repelled by the negatively charged N atoms of the tetrazole groups. In order to minimize N–O interactions, the CO2 molecule orients at an angle and tilts out-of-plane with respect to the [Cu3O(N4CR)3] trimers, leading to a less favorable interaction (Fig. 17(a)). The magnitude of the calculated binding energy for the interaction between CO2 and the Cu2+ ion of the truncated hexatopic building unit was indeed greater for rht-MOF-pyr relative to rht-MOF-1 (−22.6 vs. −21.3 kJ mol−1).
IV. Conclusion
The important experimental and theoretical studies that have yielded detailed insights into the gas sorption sites in MOFs belonging to the popular rht-MOF platform were discussed in this highlight. NPD experiments can perhaps provide the most definitive information on the sorption sites in these MOFs as demonstrated by Yan et al. on NOTT-112.13 INS studies can give hints on the locations of the sorption sites in rht-MOFs based on the transition energies and intensities of the peaks observed in the spectra.25,48 The assignments of the peaks within the INS spectra are usually tentative, but can be confirmed through calculation of the quantum dynamics within an accurate potential energy surface.47MC simulations, especially those within the grand canonical ensemble, are probably the most convenient theoretical methods used to investigate the binding sites in rht-MOFs. The sorption sites can be determined through examining the modeled structure at various loadings. Plotting the density contours, three-dimensional histograms, g(r), induced dipole distributions, and density distribution profiles from the MC simulations have also been implemented to evaluate the binding sites in rht-MOFs. It is important to note that the accuracy of such classical simulations to predict the binding sites in these materials is typically dependent on the potential energy function utilized for the MOF and sorbate. Although less commonly employed, electronic structure calculations can reveal information on the orientation and binding energies for a sorbate localized about certain components or functionalities in rht-MOFs, as done previously for rht-MOF-9 in ref. 33 and rht-MOF-1 and rht-MOF-pyr in ref. 36.
We note that this paper discussed numerous studies that elucidated the H2 and CO2 sorption mechanisms and binding sites in rht-MOFs. To the best of our knowledge, the sorption sites for other guest molecules (e.g., CH4, N2, C2 hydrocarbons) in these MOFs have not been thoroughly explored, even though experimental gas sorption measurements were performed with such sorbates.12,19,29,30,33 Nevertheless, it is likely that the binding sites for these gases in rht-MOFs are analogous to those described herein for H2 and CO2. Indeed, both of these sorbates occupied very similar sites in the structure for a particular rht-MOF.
On the basis of the studies summarized in this paper, it can be concluded that the most favorable sorbate binding sites in all neutral rht-MOFs are the exposed M2+ ions of the [M2(O2CR)4] clusters. The results from the NPD studies presented in ref. 13 clearly demonstrated that the M2+ ions that make up the metal paddlewheels in rht-MOFs are chemically distinct, and therefore exhibit different binding affinities toward the sorbates. This finding has been supported through GCMC simulations of gas sorption in various rht-MOFs.28,45,52,56,58–61 The corners of the T-Td cages have also been discovered as preferential sorption sites in these MOFs. In addition, if polar functional groups, such as amide,15 secondary amine,19,20,31,33 1,3,5-triazine,19,20 or 1,2,3-triazole27–29 are present on the organic linkers, the sorbate molecules can interact with these moieties as well, generally leading to an increase in gas uptake at low pressures. In a cationic rht-MOF containing extra-framework counterions, such rht-MOF-1, the counterions represent the most favorable sorption sites for the guest molecules in these MOFs.36,52,53 The M2+ ions that are part of the M3O trimers are also preferred binding sites in rht-MOFs possessing such clusters (e.g., rht-MOF-1 and rht-MOF-pyr).36,52 At higher loadings, sorption can be observed within the spacious regions of the three distinct cages of the rht-MOF structure.
The experimental and theoretical probing techniques described herein can provide significant understanding of the gas sorption mechanisms and binding sites in rht-MOFs and other types of MOFs in general. The results from such studies can help guide scientists and engineers to gain new perspectives into creating novel porous materials with improved gas uptake and adsorption enthalpy. After all, an ultimate goal in this energy economy is to synthesize innovative structures that can target certain environmental applications, such as addressing the challenging Department of Energy (DOE) targets for on-board H2 and CH4 storage81,82 and effectively capturing CO2 from post-combustion effluents or directly from the atmosphere to mitigate the global warming phenomenon.83
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the National Science Foundation (Award No. DMR-1607989), including support from the Major Research Instrumentation Program (Award No. CHE-1531590). B. S. also acknowledges support from an American Chemical Society Petroleum Research Fund grant (ACS PRF 56673-ND6).References
- S. L. James, Chem. Soc. Rev., 2003, 32, 276–288 RSC
.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed
- T. A. Makal, J.-R. Li, W. Lu and H.-C. Zhou, Chem. Soc. Rev., 2012, 41, 7761–7779 RSC
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed
-
M. Eddaoudi and J. F. Eubank, in Metal–Organic Frameworks: Design and Application, ed. L. R. MacGillivray, John Wiley & Sons, Inc., Hoboken, NJ, 2010, pp. 37–89 Search PubMed
- J. J. Perry IV, J. A. Perman and M. J. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400–1417 RSC
- F. Nouar, J. F. Eubank, T. Bousquet, L. Wojtas, M. J. Zaworotko and M. Eddaoudi, J. Am. Chem. Soc., 2008, 130, 1833–1835 CrossRef CAS PubMed
- Y. Zou, M. Park, S. Hong and M. S. Lah, Chem. Commun., 2008, 2340–2342 RSC
- Y. Yan, X. Lin, S. Yang, A. J. Blake, A. Dailly, N. R. Champness, P. Hubberstey and M. Schröder, Chem. Commun., 2009, 1025–1027 RSC
- S. Hong, M. Oh, M. Park, J. W. Yoon, J.-S. Chang and M. S. Lah, Chem. Commun., 2009, 5397–5399 RSC
- D. Zhao, D. Yuan, D. Sun and H.-C. Zhou, J. Am. Chem. Soc., 2009, 131, 9186–9188 CrossRef CAS PubMed
- D. Yuan, D. Zhao, D. Sun and H.-C. Zhou, Angew. Chem., 2010, 122, 5485–5489 CrossRef
- Y. Yan, I. Telepeni, S. Yang, X. Lin, W. Kockelmann, A. Dailly, A. J. Blake, W. Lewis, G. S. Walker, D. R. Allan, S. A. Barnett, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2010, 132, 4092–4094 CrossRef CAS PubMed
- O. K. Farha, A. O. Yazaydin, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944–948 CrossRef CAS PubMed
- B. Zheng, J. Bai, J. Duan, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2011, 133, 748–751 CrossRef CAS PubMed
- Y. Yan, A. J. Blake, W. Lewis, S. A. Barnett, A. Dailly, N. R. Champness and M. Schröder, Chem. – Eur. J., 2011, 17, 11162–11170 CrossRef CAS PubMed
- Y. Yan, S. Yang, A. J. Blake, W. Lewis, E. Poirier, S. A. Barnett, N. R. Champness and M. Schröder, Chem. Commun., 2011, 47, 9995–9997 RSC
- D. Yuan, D. Zhao and H.-C. Zhou, Inorg. Chem., 2011, 50, 10528–10530 CrossRef CAS PubMed
- B. Li, Z. Zhang, Y. Li, K. Yao, Y. Zhu, Z. Deng, F. Yang, X. Zhou, G. Li, H. Wu, N. Nijem, Y. J. Chabal, Z. Lai, Y. Han, Z. Shi, S. Feng and J. Li, Angew. Chem., Int. Ed., 2012, 51, 1412–1415 CrossRef CAS PubMed
- R. Luebke, J. F. Eubank, A. J. Cairns, Y. Belmabkhout, L. Wojtas and M. Eddaoudi, Chem. Commun., 2012, 48, 1455–1457 RSC
- B. Zheng, Z. Yang, J. Bai, Y. Li and S. Li, Chem. Commun., 2012, 48, 7025–7027 RSC
- I. Eryazici, O. K. Farha, B. G. Hauser, A. O. Yazaydin, A. A. Sarjeant, S. T. Nguyen and J. T. Hupp, Cryst. Growth Des., 2012, 12, 1075–1080 CAS
- O. K. Farha, C. E. Wilmer, I. Eryazici, B. G. Hauser, P. A. Parilla, K. O'Neill, A. A. Sarjeant, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 9860–9863 CrossRef CAS PubMed
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. Ö. Yazaydin and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016–15021 CrossRef CAS PubMed
- J. F. Eubank, F. Nouar, R. Luebke, A. J. Cairns, L. Wojtas, M. Alkordi, T. Bousquet, M. R. Hight, J. Eckert, J. P. Embs, P. A. Georgiev and M. Eddaoudi, Angew. Chem., Int. Ed., 2012, 51, 10099–10103 CrossRef CAS PubMed
- X. Zhao, D. Sun, S. Yuan, S. Feng, R. Cao, D. Yuan, S. Wang, J. Dou and D. Sun, Inorg. Chem., 2012, 51, 10350–10355 CrossRef CAS PubMed
- X.-J. Wang, P.-Z. Li, Y. Chen, Q. Zhang, H. Zhang, X. X. Chan, R. Ganguly, Y. Li, J. Jiang and Y. Zhao, Sci. Rep., 2013, 3, 1149 CrossRef PubMed
- Y. Yan, M. Suyetin, E. Bichoutskaia, A. J. Blake, D. R. Allan, S. A. Barnett and M. Schröder, Chem. Sci., 2013, 4, 1731–1736 RSC
- C. E. Wilmer, O. K. Farha, T. Yildirim, I. Eryazici, V. Krungleviciute, A. A. Sarjeant, R. Q. Snurr and J. T. Hupp, Energy Environ. Sci., 2013, 6, 1158–1163 CAS
- G. Barin, V. Krungleviciute, D. A. Gomez-Gualdron, A. A. Sarjeant, R. Q. Snurr, J. T. Hupp, T. Yildirim and O. K. Farha, Chem. Mater., 2014, 26, 1912–1917 CrossRef CAS
- R. Luebke, L. J. Weselinski, Y. Belmabkhout, Z. Chen, L. Wojtas and M. Eddaoudi, Cryst. Growth Des., 2014, 14, 414–418 CAS
- X.-J. Wang, J. Li, P.-Z. Li, L.-B. Xing, H. Lu, H. Wu, Y. Shi, R. Zou and Y. Zhao, Inorg. Chem. Commun., 2014, 46, 13–16 CrossRef CAS
- K. Liu, B. Li, Y. Li, X. Li, F. Yang, G. Zeng, Y. Peng, Z. Zhang, G. Li, Z. Shi, S. Feng and D. Song, Chem. Commun., 2014, 50, 5031–5033 RSC
- J. Li, P.-Z. Li, Q.-Y. Li, Y. Cao, H. Lu, H. Wu, F. Li, Y. Shi, X.-J. Wang and Y. Zhao, RSC Adv., 2014, 4, 53975–53980 RSC
- W.-Y. Gao, R. Cai, T. Pham, K. A. Forrest, A. Hogan, P. Nugent, K. Williams, L. Wojtas, R. Luebke, L. J. Weselinski, M. J. Zaworotko, B. Space, Y.-S. Chen, M. Eddaoudi, X. Shi and S. Ma, Chem. Mater., 2015, 27, 2144–2151 CrossRef CAS
- W.-Y. Gao, T. Pham, K. A. Forrest, B. Space, L. Wojtas, Y.-S. Chen and S. Ma, Chem. Commun., 2015, 51, 9636–9639 RSC
- X.-J. Wang, J. Li, Q.-Y. Li, P.-Z. Li, H. Lu, Q. Lao, R. Ni, Y. Shi and Y. Zhao, CrystEngComm, 2015, 17, 4632–4636 RSC
- Q.-Y. Li, Y. Quan, W. Wei, J. Li, H. Lu, R. Ni and X.-J. Wang, Polyhedron, 2015, 99, 1–6 CrossRef CAS
- X. Guo, Z. Zhou, C. Chen, J. Bai, C. He and C. Duan, ACS Appl. Mater. Interfaces, 2016, 8, 31746–31756 CAS
- J. Liu, G. Liu, C. Gu, W. Liu, J. Xu, B. Li and W. Wang, J. Mater. Chem. A, 2016, 4, 11630–11634 CAS
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed
- B. Chen, N. W. Ockwig, A. R. Millward, D. S. Contreras and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4745–4749 CrossRef CAS PubMed
- V. K. Peterson, Y. Liu, C. M. Brown and C. J. Kepert, J. Am. Chem. Soc., 2006, 128, 15578–15579 CrossRef CAS PubMed
- D. Franz, K. A. Forrest, T. Pham and B. Space, Cryst. Growth Des., 2016, 16, 6024–6032 CAS
-
J. Eckert and W. Lohstroh, in Neutron Applications in Materials for Energy, ed. G. J. Kearley and V. K. Peterson, Springer International Publishing, 2015, pp. 205–239 Search PubMed
- T. Pham, K. A. Forrest, B. Space and J. Eckert, Phys. Chem. Chem. Phys., 2016, 18, 17141–17158 RSC
- T. Pham, K. A. Forrest, J. Eckert, P. A. Georgiev, A. Mullen, R. Luebke, A. J. Cairns, Y. Belmabkhout, J. F. Eubank, K. McLaughlin, W. Lohstroh, M. Eddaoudi and B. Space, J. Phys. Chem. C, 2014, 118, 439–456 CAS
- S. Ma, J. Eckert, P. M. Forster, J. W. Yoon, Y. K. Hwang, J.-S. Chang, C.
D. Collier, J. B. Parise and H.-C. Zhou, J. Am. Chem. Soc., 2008, 130, 15896–15902 CrossRef CAS PubMed
- C. M. Brown, Y. Liu, T. Yildirim, V. K. Peterson and C. J. Kepert, Nanotechnology, 2009, 20, 204025 CrossRef PubMed
- X.-S. Wang, S. Ma, P. Forster, D. Yuan, J. Eckert, J. López, B. Murphy, J. Parise and H.-C. Zhou, Angew. Chem., Int. Ed., 2008, 47, 7263–7266 CrossRef CAS PubMed
- T. Pham, K. A. Forrest, A. Hogan, K. McLaughlin, J. L. Belof, J. Eckert and B. Space, J. Mater. Chem. A, 2014, 2, 2088–2100 CAS
- R. Babarao, M. Eddaoudi and J. W. Jiang, Langmuir, 2010, 26, 11196–11203 CrossRef CAS PubMed
- Y. Liu, J. F. Eubank, A. J. Cairns, J. Eckert, V. C. Kravtsov, R. Luebke and M. Eddaoudi, Angew. Chem., Int. Ed., 2007, 46, 3278–3283 CrossRef CAS PubMed
- T. Pham, K. A. Forrest, A. Hogan, B. Tudor, K. McLaughlin, J. L. Belof, J. Eckert and B. Space, Cryst. Growth Des., 2015, 15, 1460–1471 CAS
- K. A. Forrest, T. Pham, K. McLaughlin, J. L. Belof, A. C. Stern, M. J. Zaworotko and B. Space, J. Phys. Chem. C, 2012, 116, 15538–15549 CAS
- Z. Zhang, Z. Li and J. Li, Langmuir, 2012, 28, 12122–12133 CrossRef CAS PubMed
- T. Pham, K. A. Forrest, P. Nugent, Y. Belmabkhout, R. Luebke, M. Eddaoudi, M. J. Zaworotko and B. Space, J. Phys. Chem. C, 2013, 117, 9340–9354 CAS
- T. Pham, K. A. Forrest, K. McDonald and B. Space, Cryst. Growth Des., 2014, 14, 5599–5607 CAS
- K. Zhang, A. Nalaparaju and J. Jiang, J. Mater. Chem. A, 2015, 3, 16327–16336 CAS
- T. Pham, K. A. Forrest, W.-Y. Gao, S. Ma and B. Space, ChemPhysChem, 2015, 16, 3170–3179 CrossRef CAS PubMed
- N. Metropolis, A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller and E. Teller, J. Chem. Phys., 1953, 21, 1087–1092 CrossRef CAS
- A. Z. Panagiotopoulos, Mol. Phys., 1987, 61, 813–826 CrossRef CAS
- J. L. Belof, A. C. Stern and B. Space, J. Chem. Theory Comput., 2008, 4, 1332–1337 CrossRef CAS PubMed
- V. Buch, J. Chem. Phys., 1994, 100, 7610–7629 CrossRef CAS
- J. L. Belof, A. C. Stern, M. Eddaoudi and B. Space, J. Am. Chem. Soc., 2007, 129, 15202–15210 CrossRef CAS PubMed
- G. E. Scuseria, M. D. Miller, F. Jensen and J. Geertsen, J. Chem. Phys., 1991, 94, 6660–6663 CrossRef CAS
- J. J. Potoff and J. I. Siepmann, AIChE J., 2001, 47, 1676–1682 CrossRef CAS
- A. L. Mullen, T. Pham, K. A. Forrest, C. R. Cioce, K. McLaughlin and B. Space, J. Chem. Theory Comput., 2013, 9, 5421–5429 CrossRef CAS PubMed
- J.-R. Li, J. Yu, W. Lu, L.-B. Sun, J. Sculley, P. B. Balbuena and H.-C. Zhou, Nat. Commun., 2013, 4, 1538 CrossRef PubMed
- S. K. Elsaidi, M. H. Mohamed, T. Pham, T. Hussein, L. Wojtas, M. J. Zaworotko and B. Space, Cryst. Growth Des., 2016, 16, 1071–1080 CAS
- Y. Peng, V. Krungleviciute, I. Eryazici, J. T. Hupp, O. K. Farha and T. Yildirim, J. Am. Chem. Soc., 2013, 135, 11887–11894 CrossRef CAS PubMed
- F. Darkrim and D. Levesque, J. Chem. Phys., 1998, 109, 4981–4984 CrossRef CAS
- H. Wu, J. M. Simmons, G. Srinivas, W. Zhou and T. Yildirim, J. Phys. Chem. Lett., 2010, 1, 1946–1951 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian09 Revision A.02, Gaussian Inc., Wallingford, CT, 2016 Search PubMed
- A. L. Myers and J. M. Prausnitz, AIChE J., 1965, 11, 121–127 CrossRef CAS
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS
- M. Valiev, E. Bylaska, N. Govind, K. Kowalski, T. Straatsma, H. V. Dam, D. Wang, J. Nieplocha, E. Apra, T. Windus and W. de Jong, Comput. Phys. Commun., 2010, 181, 1477–1489 CrossRef CAS
- DOE Targets for Onboard Hydrogen Storage Systems for Light-Duty Vehicles, 2012, http://energy.gov/sites/prod/files/2015/01/f19/fcto_myrdd_table_onboard_h2_storage_systems_doe_targets_ldv.pdf, Accessed on March 24, 2017
- ARPA-E: MOVE Program
Overview, 2013, https://arpa-e.energy.gov/sites/default/files/documents/files/MOVE_ProgramOverview.pdf, Accessed on April 24, 2017
- D. G. Madden, H. S. Scott, A. Kumar, K.-J. Chen, R. Sanii, A. Bajpai, M. Lusi, T. Curtin, J. J. Perry and M. J. Zaworotko, Philos. Trans. R. Soc., A, 2017, 375, 20160025 CrossRef PubMed
| This journal is © The Royal Society of Chemistry 2017 |