
Photocatalytic metal–organic frameworks for organic transformations
Xiao
Yu
ab,
Le
Wang
b and
Seth M.
Cohen
 *b
*b
aDepartment of Nanoengineering, University of California, La Jolla, San Diego, California 92093, USA
bDepartment of Chemistry and Biochemistry, University of California, La Jolla, San Diego, California 92093, USA. E-mail: scohen@ucsd.edu
First published on 12th April 2017
Abstract
Metal–organic frameworks (MOFs) have attracted increasing attention for applications in heterogeneous photocatalysis. Modifications of metal nodes and organic linkers, as well as encapsulation of active species in the pores of MOFs enable the generation of photoactive materials for catalyzing organic transformations. MOF composites integrating noble metal nanoparticles or traditional photoactive semiconductors can combine the advantages of both materials, and also improve the photocatalytic performance via synergistic effects. In this Highlight, we discuss recent examples of photoactive MOFs and their use as photocatalysts for organic reactions.
 Xiao Yu | Xiao Yu received her B.S. degree in materials chemistry from Harbin Institute of Technology in China in 2013. She is now a Ph.D. candidate in Prof. Seth Cohen's laboratory at the University of California, San Diego. Her research interests are on metal–organic frameworks as catalysts. |
 Le Wang | Dr. Le Wang received his Ph.D. degree from the University of California, Riverside, where he worked in the area of materials chemistry with Prof. Pingyun Feng. He is now a postdoc in Prof. Seth Cohen's laboratory, working on metal–organic frameworks as catalysts. |
 Seth M. Cohen | Prof. Seth M. Cohen received his B.S and B.A degrees, in Chemistry and Political Science, from Stanford University in 1994. He earned his Ph.D. degree from the University of California, Berkeley in 1998, where he worked under the guidance of Prof. Kenneth N. Raymond. He has been a professor in the department of Chemistry and Biochemistry at the University of California, San Diego since 2001. His research interests are in the areas of metalloprotein inhibitors and metal–organic frameworks. |
1. Introduction
Interest in photocatalysis for organic transformations has been motivated by: 1) the ability of excited-state chemistry to greatly change the reactivity of molecules, when compared with the ground-state; 2) the mild reaction conditions under which many photochemical reactions occur without the use of additional, exogenous reagents that might be toxic; 3) the high yields and selectivity that can be achieved by avoiding thermally induced side reactions thereby diminishing the formation of byproducts.1,2 To realize the ‘green’ potential of photochemical reactions, chemists have devoted tremendous efforts to the field since the 1960s. Photochemical organic transformations have generally required the use of UV light sources because most organic molecules only absorb UV light. The use of only UV light can be limiting, because the high energy of UV photons can lead to undesired substrate decomposition or side reactions. Fortunately, due to the development of efficient visible-light activated photoorganocatalysts3–5 and transition-metal photocatalysts,6,7 there has been a resurgence of interest in organic photochemistry in the last several years. Many of the most commonly employed visible light photocatalysts are polypyridyl ruthenium(II) and iridium(III) complexes (Fig. 1), because of their ability to absorb visible light producing long-lived, stable excited states.7 These complexes can be used as single electron transfer catalysts, serving as either an oxidant or reductant during the catalytic cycle. Leading work from several groups, including Macmillan,8–10 Yoon,11–13 and Stephenson,14,15 have expanded the variety of reactions that utilize polypyridyl Ru/Ir complexes as photoredox catalysts. The reader is referred to recent reviews for more in-depth discussions of visible-light photoreactions and photocatalysts.16–18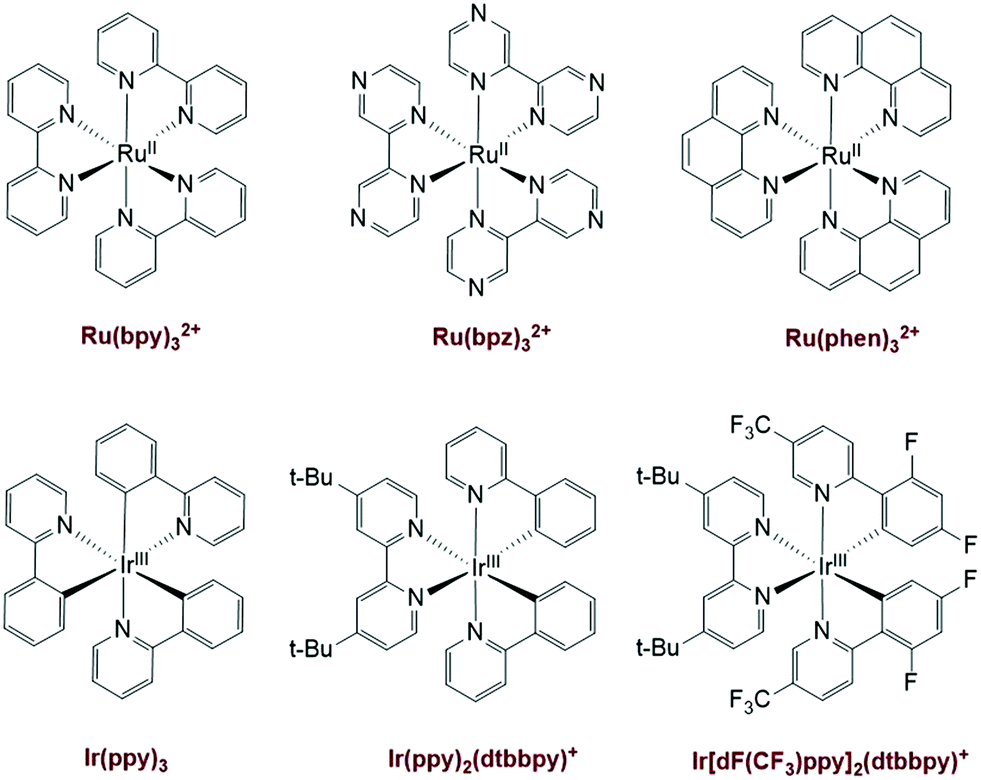 | ||
| Fig. 1 Ruthenium and iridium polypyridyl complexes that have been used as visible light photocatalysts.7 | ||
Despite these advancements, these improved homogeneous photocatalysts cannot be easily separated from the reaction mixture and recovered for further use. Therefore, to facilitate recovery/reuse of these precious metal catalysts, heterogeneous systems have been developed by immobilizing known homogeneous photocatalysts into solid-state materials including porous silica, zeolites, and polymers.19 In addition to achieving the desired heterogeneity for facile separation and recovery, it has also been found that photochemical reactions occurring inside the confined space of these materials can undergo distinct reaction pathways, leading to unprecedented reactivity and alternative reaction outcomes that are not accessible in homogeneous systems.20,21 Other attractive features of heterogeneous photocatalytic systems include improved photostability, charge separation, and enhanced photocatalytic efficiency.19
Metal–organic frameworks (MOFs), a class of porous crystalline materials constructed by inorganic metal nodes (referred to as secondary building units, SBUs) and organic linkers, hold great promises in heterogeneous photocatalysis. These materials generally possess permanent porosity that can facilitate mass transportation. In addition, the SBUs and ligands can serve as isolated catalytic centers, enhancing activity and stability.22–24 Moreover, the tunable nature and porous structure of MOF materials offer a versatile and programmable environment around the catalytic site.25 Both pre- and post-synthetic methods have been developed to modify MOFs and enhance their physical and chemical properties.26
In this Highlight, recent developments in photocatalytic MOFs for organic transformations will be discussed. Several reviews have been published on artificial photosynthesis catalyzed by MOFs, but far fewer focus on organic transformations.27–29 The modification of SBUs, incorporation of dye molecules or photoresponsive groups on the ligands, and encapsulation of photoactive species into pores will be discussed in the sections below. Photocatalytic MOF composites, including metal nanoparticles in MOFs and MOFs decorated with semiconductors are also described.
2. Photocatalysis using catalytic SBUs
The structural similarity between the SBUs in MOFs and bulk metal oxide semiconductors, which possess photocatalytic ability, has been recognized by inorganic chemists and material scientists. Therefore, efforts have been made to exploit the existing SBUs in MOFs as catalytic sites. The catalytic activity of MOF SBUs has been investigated for applications including H2 evolution, CO2 reduction, and organic pollutants degradation. Even though these applications are not the topic of this review, these studies are relevant to other efforts that utilize photocatalytic MOFs for organic transformations. Early examples of photocatalytic reactions utilizing MOF SBUs were achieved by UV light excitation. UiO-66 ([Zr6O4(OH)4(bdc)12], bdc2− = 1,4-benzenedicarboxylate) (UiO = University of Oslo), first developed by Cavka et al. in 2008,30 showed high chemical stability for the photocatalytic evolution of H2 upon UV irradiation.31 The Zr-MOF undergoes a long-lived charge separation after light excitation with electrons populating the Zr-oxo SBUs (empty metal orbitals) and holes populating the bdc2− linkers (O, C, N 2p orbitals), confirmed by spectroscopic experiments and theoretical calculations. The photocatalytic activity of stable Ti(IV)-based SBU MOFs have also been described as the corner sharing Ti-oxo octahedral of the SBUs resembles the connectivity in brookite and rutile TiO2, which is one of the most well established photocatalysts.32 In these systems, photogenerated Ti(III) could reduce molecular oxygen to form superoxide radical, which oxidizes organic molecules/pollutants.In order to employ sunlight as a sustainable light source, photocatalysis using visible light irradiation is of increasing importance. In a rare example of visible light activation of an SBU, Roeffaers and coworkers reported visible light activation of Fe-based MOFs with Fe3-μ3-oxo clusters acting as both light absorber and catalytic sites for degradation of Rhodamine 6G.33 Owing to the small size of the Fe3-μ3-oxo cluster, limited charge recombination and thus high photocatalytic activity was expected when charge carriers were enabled to effectively reach reactants on the surface. Li and coworkers employed two Fe-based MOFs, MIL-100(Fe) and MIL-68(Fe), to catalyze benzene hydroxylation to phenol with H2O2 as an oxidant using visible light.34 A conversion of ∼31% was achieved with a H2O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :benzene ratio of 3:4 with MIL-100(Fe) as catalyst for a 24 h reaction time. High selectivity (98%) was observed with no overoxidation products, such as diphenol or quinone. KIE (KIE = kinetic isotope effect) experiments, as well as EPR (EPR = electron paramagnetic resonance) spectroscopy, revealed that benzene hydroxylation to phenol proceeds via an in situ formed ˙OH radical.35 Lower conversions (14%) and selectivity (90%) were obtained for phenol with MIL-68(Fe) as the photocatalyst, showing that the structure of the MOF significantly influences the photocatalytic ability. Few other reports are available that utilize the SBUs of MOFs for visible light photocatalysis due, in part, to the poor visible light absorption for many SBUs. The poor visible light absorption of many SBUs necessitates additional modifications, such as ligand functionalization, to enhance light absorption and thereby photoactivity.
:benzene ratio of 3:4 with MIL-100(Fe) as catalyst for a 24 h reaction time. High selectivity (98%) was observed with no overoxidation products, such as diphenol or quinone. KIE (KIE = kinetic isotope effect) experiments, as well as EPR (EPR = electron paramagnetic resonance) spectroscopy, revealed that benzene hydroxylation to phenol proceeds via an in situ formed ˙OH radical.35 Lower conversions (14%) and selectivity (90%) were obtained for phenol with MIL-68(Fe) as the photocatalyst, showing that the structure of the MOF significantly influences the photocatalytic ability. Few other reports are available that utilize the SBUs of MOFs for visible light photocatalysis due, in part, to the poor visible light absorption for many SBUs. The poor visible light absorption of many SBUs necessitates additional modifications, such as ligand functionalization, to enhance light absorption and thereby photoactivity.
Amine-functionalized aromatic ligands are widely used to enable visible light absorption for photoactive MOFs. In these MOFs, SBUs still act as catalytic sites with electrons transferring from the organic ligands to the metal clusters upon visible light excitation.36 UiO-66-NH2 has been reported as an efficient visible light photocatalyst for aerobic oxidation of compounds including alcohols, olefins, and cycloalkanes.37 For example, several alcohols could be quantitatively oxidized by UiO-66-NH2 with 100% selectivity for aldehyde/ketone products. Cyclohexane was completely converted to cyclohexanone by this photocatalyst. Conversion rates decreased in the order of benzyl alcohol > cyclohexanol > hexyl alcohol > cyclohexane, depending primarily on the activation energy of α-C–H bonds. 18O isotope labelling experiment confirmed that molecular oxygen was the oxygen source in these reactions. To get insight into the mechanism of the photocatalytic process, EPR spectroscopy was carried out to monitor the intermediates formed during light irradiation. EPR indicated the formation of superoxide radical anion (O2−˙). The radical can be stabilized by interacting with the amine groups of UiO-66-NH2 and/or organic solvents, which benefits the photocatalytic oxygenations of C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. Amine functionalization of the bdc2− ligand (e.g. NH2-bdc2−) in UiO-66-NH2 moves the absorption band edge to 450 nm, unlike the unfunctionalized bdc2− analog (UiO-66), which is transparent between 350–800 nm. Indeed, UiO-66-NH2 was active for oxygenation reactions described above, while ZrO2 and UiO-66 were catalytically inactive for the same reactions.
C bonds. Amine functionalization of the bdc2− ligand (e.g. NH2-bdc2−) in UiO-66-NH2 moves the absorption band edge to 450 nm, unlike the unfunctionalized bdc2− analog (UiO-66), which is transparent between 350–800 nm. Indeed, UiO-66-NH2 was active for oxygenation reactions described above, while ZrO2 and UiO-66 were catalytically inactive for the same reactions.
3. Photocatalysis using catalytic ligands
More than any other subtopic in photocatalytic MOFs, photocatalysis using functionalized ligands has garnered the greatest attention. Organic ligands in photoactive MOFs can work as both light antennae and catalytic sites. Benefiting from well understood molecular photochemistry, various photocatalytic ligands can be easily modified for incorporation into MOFs. Among functionalized ligands, polypyridyl Ru(II) and Ir(III) ‘metalloligand’ complexes have attracted increasing attention.7,17,38 Considering the high cost of these transition metal complexes, it is of great value to develop heterogeneous and reusable analogs of these catalytic systems.In 2011, Lin and coworkers first incorporated polypyridyl Ru(II) and Ir(III) complexes into the UiO-67 (Zr6O4(OH)4(bpdc)6, bpdc2− = 4,4′-biphenyldicarboxylate) framework via a direct (mixed ligand) synthesis method.39 The resulting UiO-67 derivatives exhibited high surface areas, with ∼2–3% loading of the polypyridyl photoactive catalytic sites. Aza-Henry reactions, oxidative coupling of amines, and oxidation of sulfides were reported from these photocatalytic MOFs (Fig. 2). The MOFs showed slightly lower activity compared to homogeneous catalysts, but exhibited good yields and reusability after three catalytic runs.
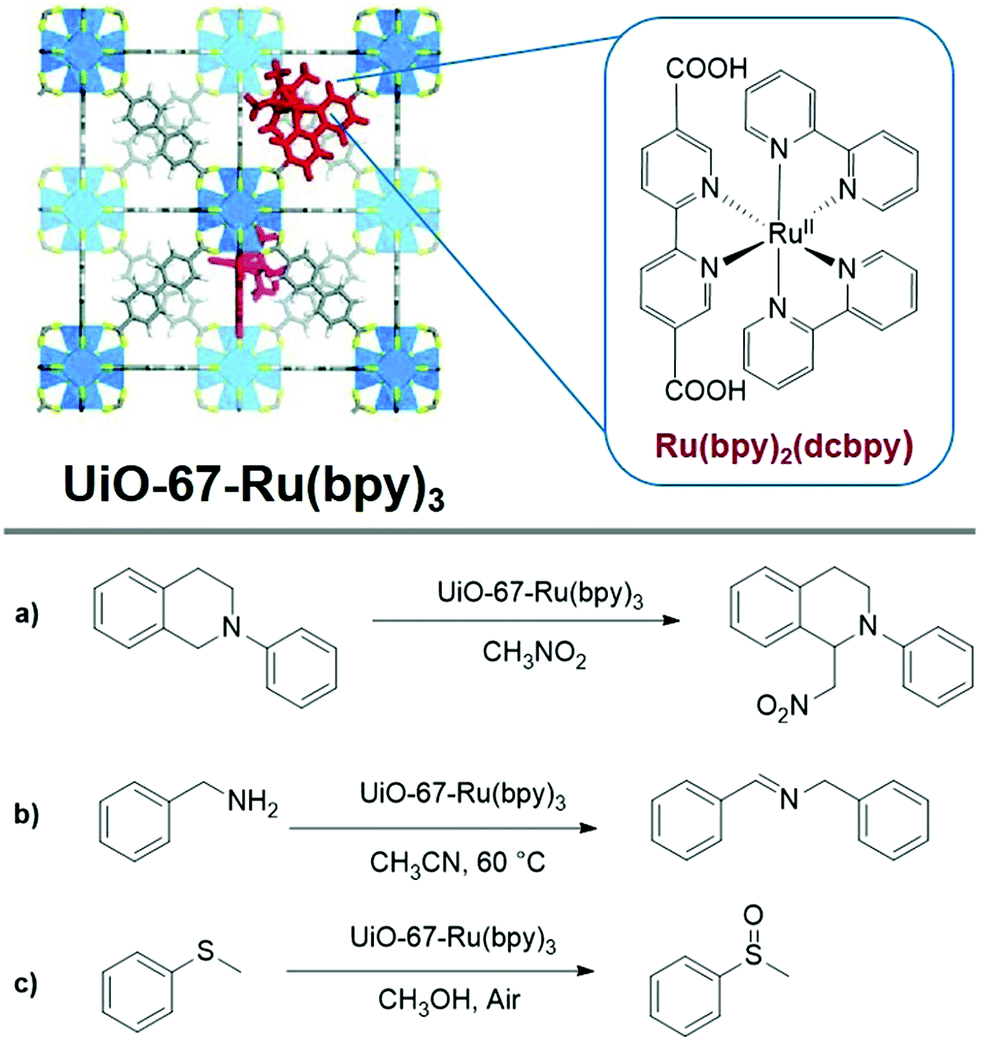 | ||
| Fig. 2 Top: Doping UiO-67 with Ru(II) polypyridyl complexes for achieving photocatalysis. Bottom: Photocatalytic reactions catalyzed by doped UiO-67 with Ru(II) polypyridyl complexes: a) Aza-Henry reactions, b) oxidative coupling of amines, and c) oxidation of sulfides. Adapted from ref. 39. | ||
Inspired by this seminal report, the Cohen laboratory incorporated [RuII(bpy)2(dcbpy)]2+ ([RuII(bpy)2(dcbpy)] = bis(2,2′-bipyridine)(5,5′-dicarboxy-2,2′ bipyridine)ruthenium(II)) (Fig. 2) into UiO-67 MOFs via postsynthetic modification (PSM), which yielded superior loading and crystallinity compared to the direct synthesis approach by Lin and coworkers.39,40 The degree of Ru incorporation could be controlled from 2% to 15% by modulating the PSM reaction time. The resulting MOFs showed stable, reusable catalytic activity for the aerobic oxidation of arylboronic acids to phenols. Similarly, [IrIII(ppy)2(dcbpy)]Cl ([IrIII(ppy)2(dcbpy)]Cl = bis(4-phenyl-2-pyridine)(5,5′-dicarboxyl-2,2′-bipyridine)iridium(III) chloride) and [IrIII(ppyF′)2(dcbpy)]Cl ([IrIII(ppyF′)2(dcbpy)]Cl = bis(2-(2,4-difluorophenyl)-5-(trifluoromethyl)pyridine)(5,5′-dicarboxyl-2,2′-bipyridine)iridium(III) chloride) were successfully incorporated into a UiO-67 framework via PSM (Fig. 3).41 To investigate the photocatalytic activity of the UiO-67-Ir MOFs, 2,2,2-trifluoroethylation of styrenes was explored, which is of importance because of the wide use of CF3-containing compounds in the pharmaceutical and agrochemical industries.42–44 Surprisingly, the MOF catalysts favored the formation of the desired hydroxytrifluoroethyl products, while suppressing dimerization of benzyl radicals that form undesirable byproducts (Fig. 3). The selectivity for the desired product was better than the related homogeneous catalyst and is likely due to confinement effects within the pores of MOF structure.
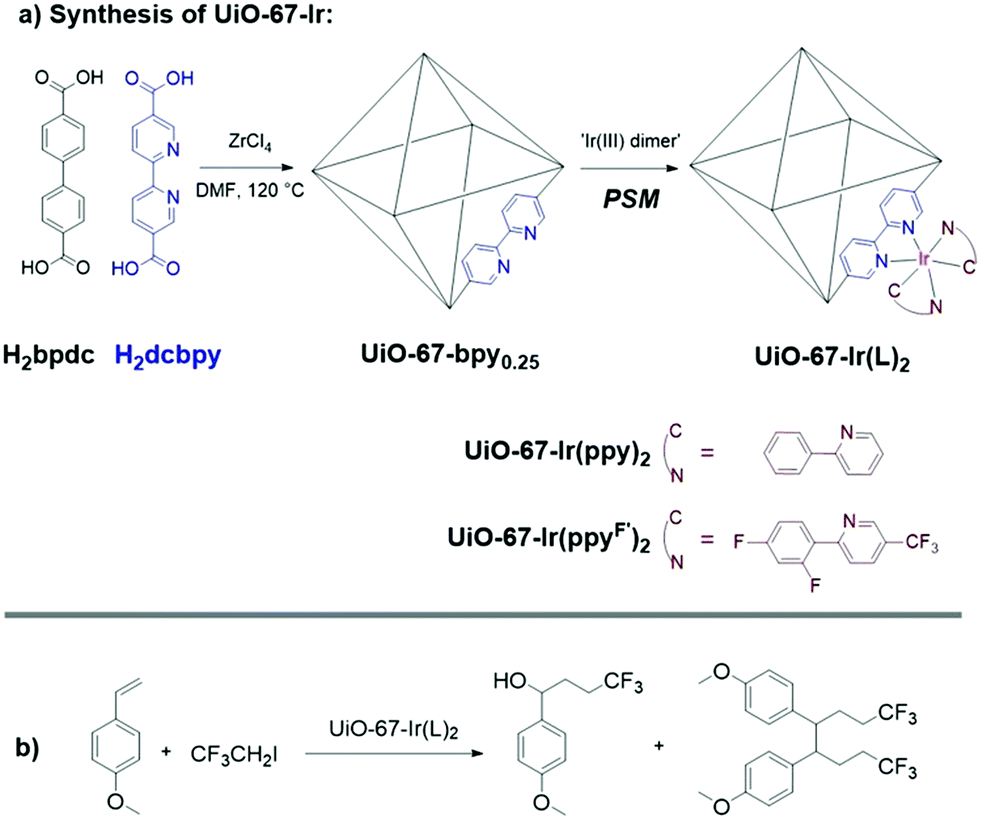 | ||
| Fig. 3 a) Synthesis of UiO-67-Ir by postsynthetic modification (PSM). b) Photocatalytic UiO-67-Ir MOFs for selective trifluoroethylation of p-methoxystyrene. | ||
In addition to Ru and Ir polypyridyl complexes, porphyrins are another important ligand used to build photocatalytic MOFs, acting as both light harvesters and photoredox centers.45,46 Porphyrin molecules typically have intense absorption bands in the visible region because of their large conjugated chromophores, and these molecules can engage in energy and/or electron transfer processes.47 Metalation of porphyrin macrocycles provides an unique handle to control optical and catalytic properties of porphyrinic MOFs. Premetalation, in situ metalation, and postsynthetic metalation are commonly-used strategies to tune the metal composition of porphyrin macrocycles in MOFs.48–50 For example, Wu and coworkers reported the synthesis of Zn–Sn-TPyP MOF (Zn–Sn-TPyP = a Sn(IV)–porphyrin-based MOF, TPyP = 5,10,15,20-tetra(4-pyridyl)-porphyrin) by using premetalated Sn–porphyrin as connecting ligand.51 The Zn-Sn-TPyP MOF worked as an efficient photocatalyst to activate molecular oxygen for the oxygenation of phenols and sulfides, showing quantitative yields for both reactions after four catalytic runs (Fig. 4). The homogeneous Sn(IV)–porphyrin analogue was largely deactivated after three catalytic runs demonstrating the importance of the MOF structure.
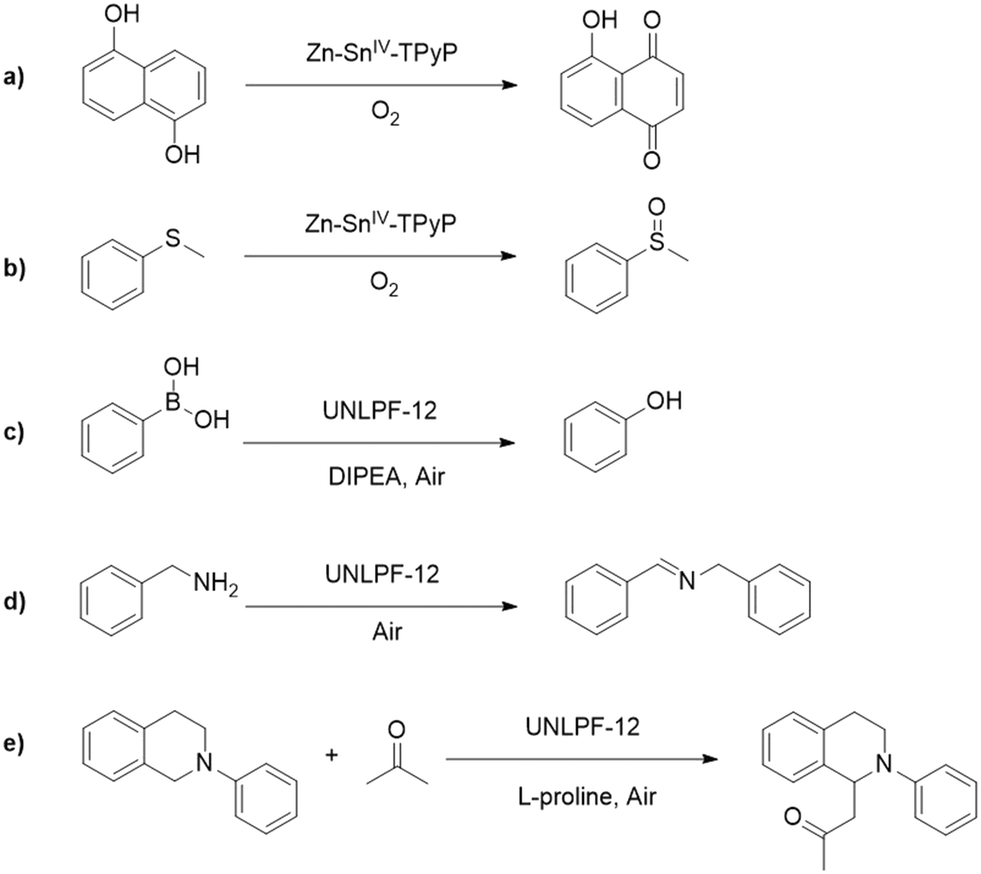 | ||
| Fig. 4 Representative reactions catalyzed by porphyrin-based MOFs: a) oxygenation of phenols catalyzed by Zn–Sn-TPyP MOF; b) oxygenation of sulphides catalyzed by Zn–Sn-TPyP MOF; c) aerobic hydroxylation of aryl boronic acids catalyzed by UNLPF-12 MOF; d) oxidative coupling of amines catalyzed by UNLPF-12 MOF; and e) Mannich reaction catalyzed by UNLPF-12. | ||
In another report, the Zhang laboratory developed a controllable synthesis of an anionic indium porphyrinic-MOF, UNLPF-10 (UNLPF = University of Nebraska-Lincoln porous framework) metalated via in situ metalation.52 The extent of metalation was tuned by varying the indium/ligand ratio in the starting solution, with H10tbcppp (H10tbcppp = tetrakis 3,5-bis[(4-carboxy)-phenyl]phenylporphyrin) as the connecting ligand. Notably, UNLPF-10 exhibited channel-like pores along all three crystal axes, which is appealing for catalysis because such pores likely provide better mass diffusion. Oxygenation of sulfide was explored as model reaction to investigate the photocatalytic ability of UNLPF-10. The rate of photo-oxygenation of thioanisole with this MOF increased as the ratio of metalation increased. In a follow-up study, four isostructural porphyrinic MOFs (namely, UNLPF-10a, -10b, -11, and -12, composed of free base, In3+–, Sn4+Cl2–, and Sn4+–porphyrin building blocks, respectively) were synthesized via postsynthetic metalation and their photocatalytic ability was investigated for three organic reactions, including the aerobic hydroxylation of arylboronic acids, oxidative primary amine coupling, and the Mannich reaction (Fig. 4).53 Compared to homogeneous catalysts, porphyrinic MOFs showed higher efficiency because of a more positive E1/2(*M/M−) (describing the half reaction *M + e− → M−, “*” denotes the excited state), which was confirmed by cyclic voltammetry. Accordingly, The E1/2 (*M/M−) of four porphyrinic MOFs were calculated to be +0.79 V, +1.25 V, +1.33 V, and +1.42 V for UNLPF-10a, -10b, -11, and -12, respectively. For the hydroxylation of 4-formylbenzeneboronic acid, UNLPF-10a gave an 87% conversion after 24 h. UNLPF-10b and -11 exhibited faster reaction rates, finishing the hydroxylation within 4 and 3.5 h. Remarkably, UNLPF-12 reached full conversion within only 2.5 h. The porphyrinic MOFs also exhibited greater photostability, resisting decomposition during photocatalytic reactions. All these studies illustrate the excellent light harvesting capability and catalytic activity of porphyrinic-MOFs showing that the nature of the central metal ion in the porphyrin plays an important role in tuning the photocatalytic behavior of porphyrinic-MOFs.
In addition to transition metal complexes and metalated porphyrins, organic chromophores (i.e. photoorganocatalysts) play an important role in photocatalysis.3,54 Recent studies on photoorganocatalysis show that most organic photocatalysts can strongly absorb visible light.55–57 Various reactions have been investigated in homogeneous photoorganocatalysis, including oxidations, reductions, alkylations and other C–C bond forming reactions, and arylations. An impressive example of photoorganocatalysis using a MOF was recently reported by Wang and coworkers,58 where a TPE-conjugated (TPE = tetraphenylethene) terphenyldicarboxylate strut was successfully incorporated into a robust UiO-68-based framework via a direct synthesis, using a mixed ligand method. The TPE-based MOF exhibited efficient photocatalytic activity for aerobic cross-dehydrogenative coupling (CDC) reactions between tertiary amines and different carbon nucleophiles mediated by visible light. No product was detected when the reaction was conducted under N2 atmosphere, indicating the importance of oxygen in the aerobic CDC reaction. EPR spectroscopy showed that O2−˙ was the active oxygen species in the photocatalytic process.
Combination of photoorganocatalysts with other catalysts in MOFs, such as enantioselective catalysts, can enrich MOF photocatalysis for more sophisticated reactions. In 2012, Duan and coworkers incorporated an organic photosensitizer and an asymmetric catalyst into the same MOF, leading to stereoselective photocatalysis.59 Two new chiral MOFs, Zn-PYI1 and Zn-PYI2, were constructed from L- or D-PYI (PYI = pyrrolidine-2-ylimidazole), and photoactive H3tca (H3tca = 4,4′,4′′-tricarboxyltriphenylamine) as connecting ligands, with Zn(II) SBUs (Fig. 5). A protected chiral ligand (L-BCIP = L-N-tertbutoxycarbonyl-2-(imidazole)-1-pyrrolidine) was used in the MOF synthesis to give Zn-PYI1 after postsynthetic deprotection (PSD). Retention of chirality was confirmed by SXRD (SXRD = single crystal X-ray crystallography) and CD spectroscopy (CD = circular dichroism). Upon visible light excitation, the strong reductive excited state of tca3− ligand induced electron transfer and provided active intermediate for α-alkylation of aldehydes, while the chiral PYI moiety acted as asymmetric catalyst to drive reactions to occur with excellent enatioselectivities. Control experiments were conducted to investigate the catalytic activity of Zn-PYI1 and Zn-PYI2, using achiral MOFs Ho-tca and MOF-150, both of which contain the same photoactive tca3− moiety. In the absence of chiral PYI, negligible alkylation product was observed for Ho-tca and MOF-150, proving that the PYI is an active component for α-alkylation of aldehydes. With additional chiral PYI molecules adsorbed into the achiral MOFs, Ho-tca catalyzed the alkylation reaction to produce the desired products in similar yields to Zn-PYI, while MOF-150 gave better yields than Zn-PYI. However, much lower enatioselectivies were observed for both Ho-tca and MOF-150 catalytic systems. The results show that the combination of both photoactive and chiral groups in a single structure worked together to achieve high activity and enantioselectivity.
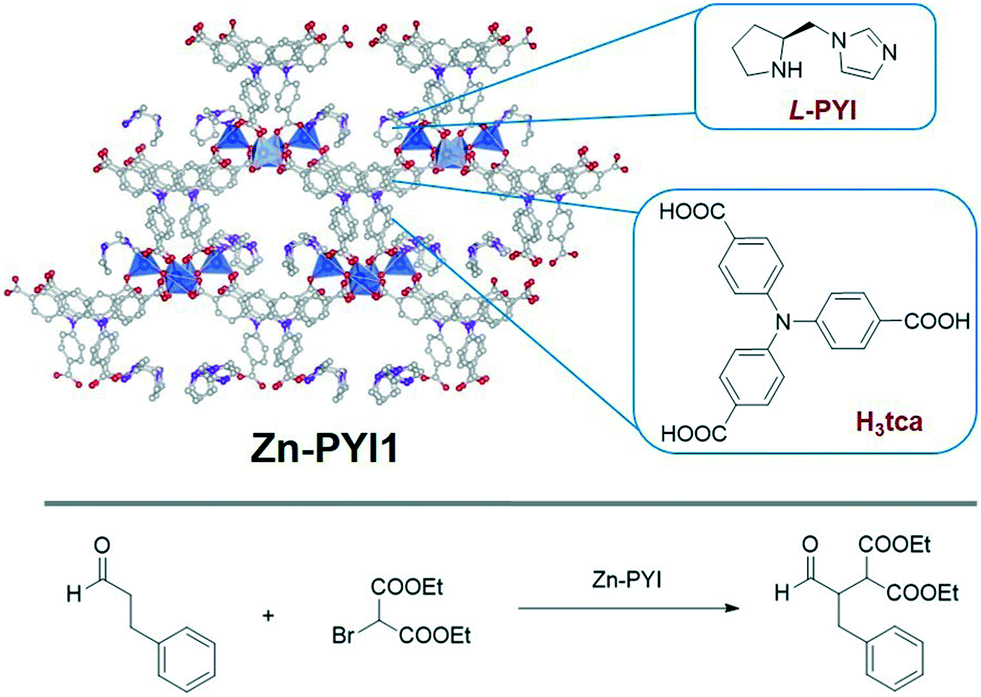 | ||
| Fig. 5 Top: Structure of chiral photoactive Zn-PYI1. The grey, red, and purple balls represent C, O, N atoms, blue polyhedra represent Zn SBUs. Bottom: Photocatalytic α-alkylation of aldehydes catalyzed by Zn-PYI MOF.59 | ||
The aforementioned photocatalysts are limited to single photon excitation and charge transfer processes. The energy of blue photons (440 nm, 2.8 eV) defines the maximum theoretical energy threshold that common visible light photocatalysts can utilize. In any system, part of the energy will be lost due to inevitable intersystem crossing and reorganization of photocatalyst excited state. The available energy of visible light photocatalysts is sufficient to reach the reduction potential of aryl iodides, but not other substrates that are more difficult to reduce such as aryl chlorides. Visible light photocatalysis employing the energies of multiple photons in one catalytic cycle are highly desirable for achieving more difficult transformations like activation of aryl chloride. The consecutive photoinduced electron transfer (conPET) process was first reported by König in 2014, opening a new door for photocatalytic conversion of less reactive chemical bonds by overcoming the energetic limitation of visible light photoredox catalysis.60 Along these lines, Duan and coworkers integrated a PDI motif (PDI = perylene diimide) into a Zn-MOF (termed Zn-PDI) achieving conPET for the visible-light-driven reduction of aryl halides (Fig. 6).61 Each PDI ligand bridged between two Zn2+ ions to form a 2D network. Strong π⋯π interaction between PDI ligands was found to group triads of PDI ligands to form J-aggregates. J-aggregates, named after Jelley who first observed the phenomenon, describe dye aggregates with a narrow absorption band that is shifted to a longer wavelength with respect to the monomer absorption band and a nearly resonant, narrow band fluorescence (very small Stokes shift).62 In this case, the formation of J-aggregates by PDI triads in Zn-PDI aids the photoactivity because of an enhanced ability to delocalize and migrate excitons. Zn-PDI reduced 4′-bromoacetophenone to acetophenone in 87% yield after only 1 h, which is superior to its homogeneous analogue (Fig. 6). Applying Zn-PDI for C–C bond formation using N-methyl pyrrole as trapping agent, exhibiting good to excellent yields for various substrates (Fig. 6). Zn-PDI also showed photoactivity for the efficient oxidation of benzylalcohols and benzylamines under mild reaction conditions using dioxygen as the oxidizing agent (Fig. 6). The photooxidation reactions were optimized to perform in acetonitrile (MeCN) with 0.1 mol% dry Zn-PDI to give good yields (76% for benzyl alcohol and 74% for phenylmethanamin). Oxidation reactions with simple Zn salts and H2PDI or a simple mixture of the two produced negligible products, which demonstrates the importance of the MOF structure. Specifically, it was noted that strong π⋯π interactions between PDIs in Zn-PDI distort the tetrahedral geometry of Zn(II) in the MOF resulting in a labile coordinated water molecule at each Zn(II) center. This suggested that the Zn(II) ions have an accessible coordination site for binding to and activating substrate molecules.
 | ||
| Fig. 6 Top: Structure of the Zn-PDI photocatalytic MOF. The grey, red, purple balls represent C, O, N atoms. Blue polyhedra represent Zn SBUs. Bottom: Photocatalytic reactions catalyzed by Zn-PDI: a) reduction of aryl halides via the conPET process; b) C–C formation by coupling reaction between aryl halides and N-methyl pyrrole via conPET process; c) photooxidation of benzyl alcohol; and d) oxidative coupling of amines.61 | ||
4. Photocatalysis using encapsulated catalysts
Rather than exploiting SBUs or ligand sites, the porous structure of MOFs can also allow for photoredox species to be encapsulated within the pores of MOFs. Isolation of these active species within the pores can serve to prevent deactivation via catalyst aggregation. In addition to being included as part of a linker, the molecular photocatalyst Ru(bpy)32+ has been immobilized in MOFs via entrapment.63,64 Zhou and coworkers reported the encapsulation of Ru(bpy)32+ into an anionic MOF via postsynthetic ion exchange.65 The anionic MOF, PCN-99, was synthesized by solvothermal reaction of In(NO3)2·xH2O with H3dcta (H3dcta = 10,15-dihydro-5H-diindolo-[3,2-a:3′,2′-c]carbazole-3,8,13-tricarboxylate acid) followed by immersion of the MOF in a solution of Ru(bpy)32+ to produce the desired photocatalytic MOF, Ru(bpy)3@PCN-99. The photocatalytic ability of Ru(bpy)3@PCN-99 was examined for oxidative hydroxylation of arylboronic acids, showing relatively high efficiency.Other fascinating studies using encapsulated photocatalysis were reported using polyoxometalates (POMs) trapped in the pores of a MOF. POMs are a class of anionic metal oxide clusters with unique redox catalytic properties.66,67 Encapsulation of POMs into the pores of MOFs has been reported for multielectron processes or thermal reactions,68,69 but the photocatalytic application of these materials has remained largely unexplored.70–72 In 2014, Duan and coworkers synthesized a POM-encapsulating MOF, CR-BPY1 (CR-BPY1 = copper/ruthenium bipyridine MOF), by incorporating the POM-based photoredox catalyst [SiW11O39Ru(H2O)]5− into the copper-based MOF pores. CR-BPY1 was active for the photocatalytic oxidative coupling of sp3 C–H bonds with oxygen as the oxidant, employing N-phenyl-tetrahydroisoquinoline and nitromethane as coupling partners under visible light irradiation (Fig. 7). The same group also reported a photosensitizing decatungstate-based MOF with 1D channels as an efficient heterogeneous catalyst for the selective C–H alkylation of aliphatic nitriles.73 The novel MOF, DT-BPY ([Cu4(BPY)6Cl2(W10O32)]·3H2O, BPY = 4,4′-bipyridine), was embedded with free [W10O32]4− in the pores, exhibiting absorption <400 nm from oxygen-to-tungsten charge transfer transitions of the POMs and absorption at 626 nm corresponding to nitrogen-to-copper charge transfer transitions of the Cu-BPY MOF. The MOF exhibits remarkable photocatalytic activity for β- or γ-site C–H alkylation of aliphatic nitriles under mild conditions, with good size-selectivity and recyclability (Fig. 7). Interestingly, the presence of a reaction occuring in these systems is visibly detectable by the color change from green to blue, which is characteristic of the H+[DT-BPY]− produced during catalysis process when the reactive excited state [DT-BPY]* abstracts a hydrogen from the β- or γ-C–H bond of aliphatic nitriles to form alkyl radicals.
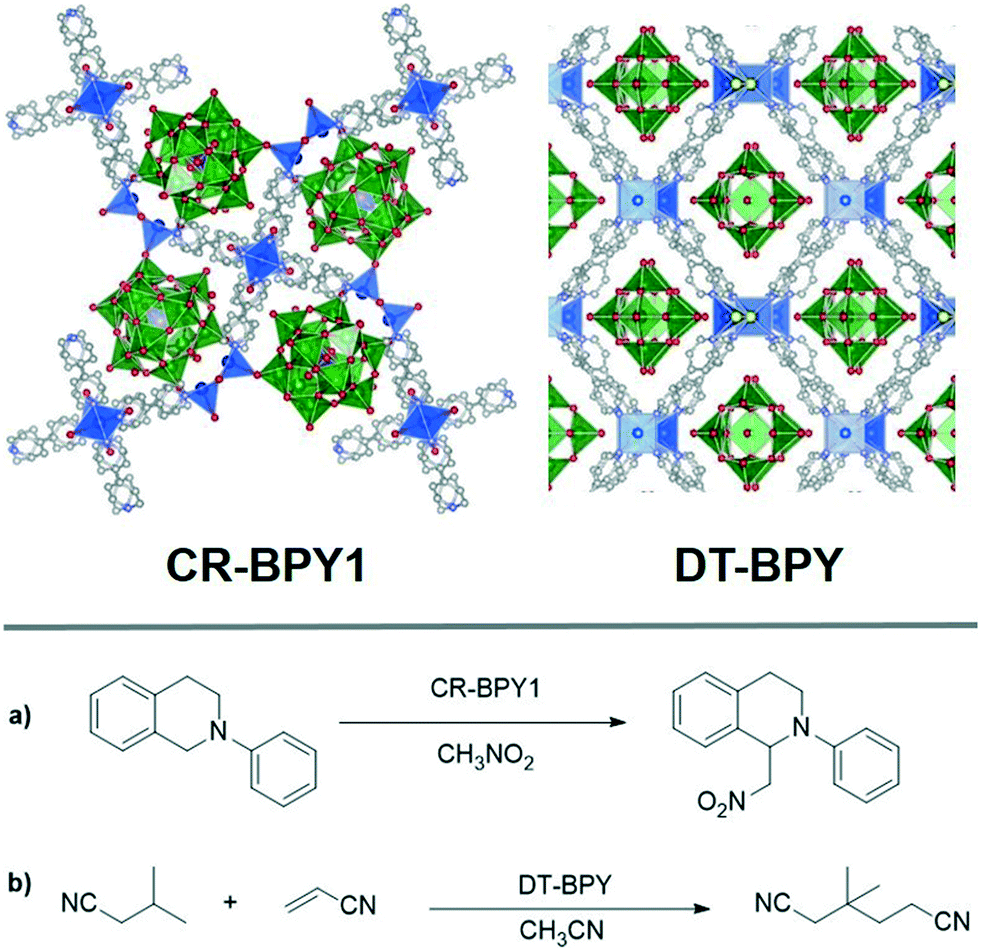 | ||
| Fig. 7 Top: Structures of POMs encapsulated MOFs CR-BPY1 and DT-BPY. The grey, red, purple, yellow balls represent C, O, N, Cl atoms. Blue and green polyhedra represent Cu SBUs and POMs, respectively. Bottom: Photocatalytic reactions catalyzed by POMs encapsulated in MOFs: a) Aza-Henry reaction induced by [SiW11O39Ru(H2O)]5− encapsulated MOF CR-BPY1 and b) selective C–H alkylation of aliphatic nitriles catalyzed by [W10O32]4− encapsulated MOF DT-BPY.73 | ||
5. Photocatalysis using MOF composites
Integration of MOFs and functional materials to form new multifunctional composites has become a rapidly developing research area. This strategy provides new opportunities to fabricate high-performance materials that can combine the merits of both components. Oxidation of benzyl alcohol is commonly exploited as model reaction to investigate the photocatalytic ability of these composites. Table 1 summarizes the various multifunctional catalytic systems described throughout this section.| Entry | Composite | Reaction conditionsa | Conv/sel (%) | Ref. |
|---|---|---|---|---|
| a O2 was used for all the reactions, benzyl alcohol was the substrate, except where noted. b 1-Benzylethanol as substrate. c Commercial CdS. d Synthesized CdS nanoparticle. e 4-Methylbenzyl alcohol as substrate. | ||||
| 1 | Au/SiO2 | 12 h, λ > 400b nm | 57/99 | 82 |
| 2 | Au@ZIF-8 multi-core | 24 h, λ > 400b nm | 52/99 | 82 |
| 3 | MIL-125(Ti) | 4 h, λ = 320–780 nm | 19/99 | 85 |
| 4 | Au/MIL-125(Ti) | 4 h, λ = 320–780 nm | 36/99 | 85 |
| 5 | Pd/MIL-125(Ti) | 4 h, λ = 320–780 nm | 33/99 | 85 |
| 6 | Pt/MIL-125(Ti) | 4 h, λ = 320–780 nm | 26/99 | 85 |
| 7 | UiO-66-NH2 | 4 h, λ > 420 nm | 20/99 | 89 |
| 8 | CdSc | 4 h, λ > 420 nm | 8/99 | 89 |
| 9 | UiO-66-NH2 + CdS | 4 h, λ > 420 nm | 18/95 | 89 |
| 10 | CdS-UiO-66-NH2 | 4 h, λ > 420 nm | 30/99 | 89 |
| 11 | CdSd | 5 h, λ > 420 nm | 40/95 | 90 |
| 12 | MIL-100(Fe) | 5 h, λ > 420 nm | 3/100 | 90 |
| 13 | CdS-MIL-100(Fe) | 5 h, λ > 420 nm | 54/99 | 90 |
| 14 | Amorphous TiO2 | 7 h, sunlighte | 6/— | 91 |
| 15 | TiO2/HKUST-1 | 7 h, sunlighte | 89/95 | 91 |
Noble metal nanoparticles (NPs) can absorb visible light due to their localized surface plasmon resonance (LSPR), which is tunable by varying the NP size, shape, and surrounding environment.74,75 In addition, NPs can also serve as electron traps and active reaction sites. These factors make noble metal NPs candidates as photocatalysts.76 Loading metal NPs into MOFs can enhance charge separation by driving photoexcited electrons from the MOF to the NPs, thus improving photocatalytic efficiency.77–81 In one example of such a hybrid system, Duan and coworkers synthesized Au@ZIF-8 core–shell structures for photocatalysis.82 Controlled synthesis of Au@ZIF-8 nanostructures was achieved by epitaxial growth or coalescence of nuclei with PVP-Au (PVP = polyvinylpyrrolidone) nanoparticles as the nucleation seeds, producing 48% single-core and 45% double- and triple-core structures (Fig. 8). The LSPR-induced light absorption of single-core Au@ZIF-8 is ∼530 nm, while multi-core particles are shifted to ∼540 nm. Photocatalytic oxidation of 1-phenylethanol was performed using resulting MOF composite under a Xe lamp with a cutoff filter (λ > 400 nm) (Fig. 8). Single-core and multi-core Au@ZIF-8 catalyzed the oxidation reaction with ∼26% and ∼52% conversion, respectively. The conversion difference between the single- and multi-core particles may be due to plasmonic coupling between Au NPs in multi-core structures. With respect to reaction selectivity, >99% selectivity was achieved for the ketone product in both cases. When compared to Au-SiO2 hybrids (∼57% yield with 99% selectivity), the lower conversion of Au@ZIF-8 could be attributed to the small pore size of ZIF-8, which limited the diffusion of the benzyl alcohol substrate.
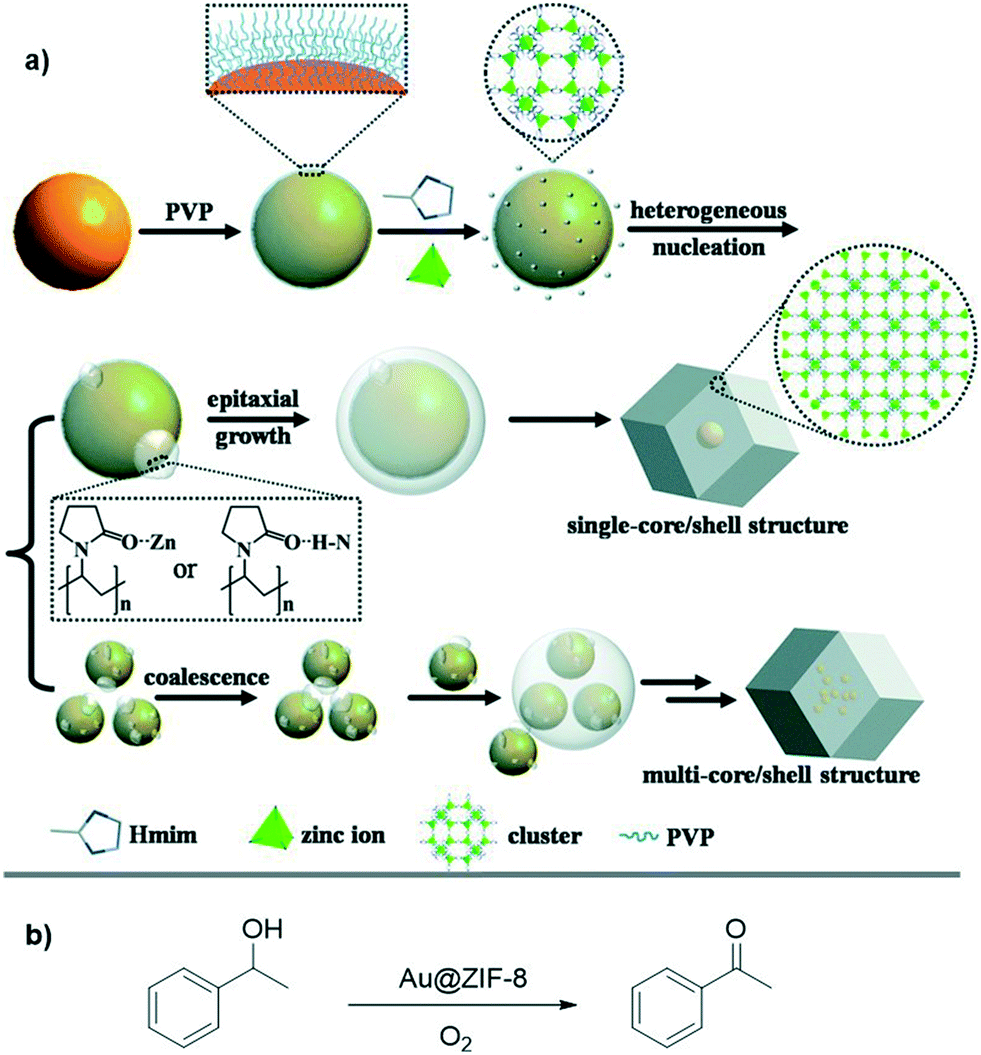 | ||
| Fig. 8 a) Schematic illustrating encapsulation process of Au NPs with ZIF-8 to form single- or multi-core–shell structures. b) Photocatalytic oxidation reaction of aryl alcohol using Au@ZIF-8 multifunctional composites. Adapted from ref. 82. | ||
In another example, Jiang and coworkers reported the encapsulation of Pd nanocubes (NCs) in ZIF-8 to obtain a composite material that was used for the efficient and selective catalytic hydrogenation of olefins at room temperature under visible light irradiation.83 The core–shell Pd-NCs@ZIF-8 composite was synthesized by introducing already-prepared Pd-NCs into a solution containing ZIF-8 precursors at 4 °C. After an hour incubation time, the Pd-NCs@ZIF-8 hybrids were 250–300 nm in size with ∼17 nm Pd-NCs inside. The Pd-NCs@ZIF-8 hybrids display a plasmonic band covering a broad UV-to-visible spectral range. Full-spectrum light irradiation was employed to promote surface-plasmon-driven photothermal hydrogenation of olefins at room temperature, which displayed greater efficiency than thermally driven catalytic hydrogenation at 50 °C. During the catalysis, the Pd-NCs@ZIF-8 composite prevented aggregation of the Pd-NCs, thereby stabilizing the Pd-NC catalyst and allowing for excellent recyclability for three successive runs with near quantitative yields, whereas with the Pd NCs alone, the yield gradually decreased to 21% by the third run. In addition, the well-defined pore structure of ZIF-8 also elicited the expected size-selective catalysis.
In addition to photocatalytically inactive MOFs like ZIF-8, the combination of noble metal NPs with photoactive MOFs can dramatically enhance the activity. For example, Matsuoka and coworkers84 deposited Pt, as confirmed by PXRD and N2 adsorption, onto a Ti-MOF-NH2. Ti-MOF-NH2 is a derivative of MIL-125(Ti), which was synthesized from TPOT (TPOT = tetrapropyl orthotitanate) and H2BDC-NH2 (H2BDC-NH2 = 2-amino-benzenedicarboxylic acid) under solvothermal conditions. Photodeposition methods were employed to prepare Pt/Ti-MOF-NH2, using H2PtCl6 as precursor. The resulting Pt/Ti-MOF-NH2 hybrid was found to photocatalytically reduce nitrobenzene under visible light irradiation, with TEOA as sacrificial agent. Nitrosobenzene was observed as an intermediate in the reaction. Control experiments showed that each feature of the hybrid was essential. That is, a coordination network of Ti-oxo clusters linked by NH2-bdc2− linkers were key factors for catalytic activity, while the Pt deposition played an important role in improving the photocatalytic performance as a cocatalyst. The wavelength dependence of the quantum efficiency for Pt/Ti-MOF-NH2 was investigated, revealing that light absorption occurs at the NH2-bdc2− linkers, followed by electron transfer to the catalytically active Ti-oxo clusters with deposited Pt. In a related study, a general approach to synthesize M/MIL-125(Ti) (M = Au, Pd and Pt NPs) composites85 resulted in noble metal NP formation directed by an in situ redox reaction. The photocatalytic activity of M/MIL-125(Ti) composites was examined for the oxidation of benzyl alcohol to benzaldehyde, displaying improved conversion and high selectivity compared to pristine MIL-125(Ti).
In addition to noble metal NPs, semiconductors have been studied for decades for photocatalytic applications.86–88 In one example of combining semiconductor NPs with MOFs, CdS nanorods were deposited on the surface of UiO-66-NH2via a room-temperature photodeposition technique.89 The CdS-UiO-66-NH2 nanocomposites display good conversion and high selectivity for photocatalytic oxidation of alcohols to the corresponding aldehydes under mild conditions. Similarly, CdS-MIL-100(Fe) nanocomposites were also reported to be an efficient photocatalyst for selective oxidation of benzyl alcohol to benzaldehyde.90
Other photocatalytic reactions have been demonstrated by decorating mesoporous HKUST-1 with amorphous TiO2 layers.91 A structure-directing agent (poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) triblock copolymer template, Pluronic P123) was used for the synthesis of HKUST-1 to produce this MOF with ordered mesoporous domains (confirmed by TEM analysis). The surface of this mesoporous HKUST-1, was decorated with amorphous TiO2 through a layer-by-layer coating. To investigate the photocatalytic performance of the TiO2-MOF composite, the aerobic oxidation of 4-methylbenzyl alcohol was tested with solar irradiation, giving 89% conversion and 95% selectivity for the partial oxidation product, i.e. 4-methylbenzylaldehyde. A variety of primary and secondary benzylic alcohols were examined, showing a reasonable substrate scope with low to high photocatalytic yields (32–100%).
6. Conclusions
Significant progress has been made in producing visible light activated photocatalytic MOFs. In this Highlight, several approaches to functionalize MOFs for photoactivity via the SBUs, organic linkers, and guest encapsulation within the MOF pores has been discussed. Among these approaches, modification of the ligand component, to extend the absorption band, is a very effective strategy due, to the variety of functional groups and dyes that can be incorporated as part of the ligand. Furthermore, combining modification methods can be used to introduce more than one functional group into the MOF for cooperative and synergistic catalysis. Many MOF photocatalysts display substrate size selectivity, due to the restricted pore aperture of the MOF, which suggests that catalysis primarily occurs within the pores of the MOF. Isolation of the photoactive components within the MOF can also result in better charge separation, reduced charge recombination, ultimately leading to improved photocatalytic efficiency when compared to homogeneous analogues.Further advancements in photocatalytic MOFs will require improvements in several aspects of these systems. First, few mechanistic studies on these photocatalytic processes have been reported, which are essential to understanding charge separation and the temporal evolution of these processes during photocatalysis. Second, MOFs with high stability under a variety of reaction conditions are needed to exploit a wider scope of organic transformations. Recent examples of stable photocatalytic MOFs rely on Zr(IV)-, Al(III)-, and Ti(IV)-based MOFs.31,39,92,93 Development of photocatalytic MOFs with different SBUs for improved redox and photoactivity will provide more opportunities for catalysis. Third, compared to the wide variety of organic reactions catalyzed by homogeneous photoredox catalysts, the scope of organic transformations performed with photocatalytic MOFs has been surprisingly limited. More high value reactions must be investigated by taking full advantage of the exciting advancements in modern photoredox chemistry. We believe that the field of photocatalytic MOFs will continue to attract attention from organic, inorganic, and physical chemists, as new advancements are made in the years to come.
Acknowledgements
Studies on the synthesis, modification, and catalytic properties of MOFs for organic transformations were supported by a grant from the National Science Foundation under Award No. CHE-1359906. Other studies in our laboratory on MOFs have been supported by grants from the Office of Basic Energy Sciences, Department of Energy under Award No. DE-FG02-08ER46519 and the Department of Defense by the Army Research Office under Award No. W911NF-16-2-0106. The authors thank Xiaoping Zhang, Jessica C. Moreton, and Sergio Ayala (U.C. San Diego) for careful reading and editing of this manuscript.Notes and references
- N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS PubMed
.
- M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725–2756 CrossRef CAS PubMed
- D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97–113 RSC
- D. W. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS PubMed
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed
- C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed
- D. A. Nicewicz and D. W. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed
- D. A. Nagib and D. W. MacMillan, Nature, 2011, 480, 224–228 CrossRef CAS PubMed
- A. McNally, C. K. Prier and D. W. MacMillan, Science, 2011, 334, 1114–1117 CrossRef CAS PubMed
- M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed
- J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, Science, 2014, 344, 392–396 CrossRef CAS PubMed
- J. M. Narayanam, J. W. Tucker and C. R. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed
- J. M. Narayanam and C. R. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC
- J. Xuan and W. J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed
- M. H. Shaw, J. Twilton and D. W. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef CAS PubMed
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed
- A. Corma and H. Garcia, Chem. Commun., 2004, 1443–1459 RSC
- V. Ramamurthy, J. Photochem. Photobiol., C, 2000, 1, 145–166 CrossRef CAS
-
V. Ramamurthy, Photochemistry in organized and constrained media, VCH publishers, 1991 Search PubMed
- S. L. James, Chem. Soc. Rev., 2003, 32, 276–288 RSC
- J. L. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef CAS
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed
- S. M. Cohen, Z. Zhang and J. A. Boissonnault, Inorg. Chem., 2016, 55, 7281–7290 CrossRef CAS PubMed
- S. M. Cohen, Chem. Rev., 2011, 112, 970–1000 CrossRef PubMed
- A. Dhakshinamoorthy, A. M. Asiri and H. García, Angew. Chem., Int. Ed., 2016, 55, 5414–5445 CrossRef CAS PubMed
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC
- M. Nasalevich, M. Van der Veen, F. Kapteijn and J. Gascon, CrystEngComm, 2014, 16, 4919–4926 RSC
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed
- C. Gomes Silva, I. Luz, F. X. Llabrés i Xamena, A. Corma and H. García, Chem. - Eur. J., 2010, 16, 11133–11138 CrossRef PubMed
- M. Dan-Hardi, C. Serre, T. Frot, L. Rozes, G. Maurin, C. Sanchez and G. Férey, J. Am. Chem. Soc., 2009, 131, 10857–10859 CrossRef CAS PubMed
- K. G. Laurier, F. Vermoortele, R. Ameloot, D. E. De Vos, J. Hofkens and M. B. Roeffaers, J. Am. Chem. Soc., 2013, 135, 14488–14491 CrossRef CAS PubMed
- D. Wang, M. Wang and Z. Li, ACS Catal., 2015, 5, 6852–6857 CrossRef CAS
- E. Brillas, I. Sirés and M. A. Oturan, Chem. Rev., 2009, 109, 6570–6631 CrossRef CAS PubMed
- D. Sun, L. Ye and Z. Li, Appl. Catal., B, 2015, 164, 428–432 CrossRef CAS
- J. Long, S. Wang, Z. Ding, S. Wang, Y. Zhou, L. Huang and X. Wang, Chem. Commun., 2012, 48, 11656–11658 RSC
- S. J. Garibay, J. R. Stork and S. M. Cohen, Prog. Inorg. Chem., 2009, 56, 335–378 CrossRef CAS
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed
- X. Yu and S. M. Cohen, Chem. Commun., 2015, 51, 9880–9883 RSC
- X. Yu and S. M. Cohen, J. Am. Chem. Soc., 2016, 138, 12320–12323 CrossRef CAS PubMed
- V. Gouverneur and K. Seppelt, Chem. Rev., 2015, 115, 563–565 CrossRef CAS PubMed
- N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC
- W.-Y. Gao, M. Chrzanowski and S. Ma, Chem. Soc. Rev., 2014, 43, 5841–5866 RSC
- S. Yuan, T.-F. Liu, D. Feng, J. Tian, K. Wang, J. Qin, Q. Zhang, Y.-P. Chen, M. Bosch and L. Zou, Chem. Sci., 2015, 6, 3926–3930 RSC
-
K. M. Kadish, K. M. Smith and R. Guilard, The Porphyrin Handbook: Inorganic, organometallic and coordination chemistry, Elsevier, 2000 Search PubMed
- D. Feng, Z. Y. Gu, J. R. Li, H. L. Jiang, Z. Wei and H. C. Zhou, Angew. Chem., 2012, 124, 10453–10456 CrossRef
- C. Y. Lee, O. K. Farha, B. J. Hong, A. A. Sarjeant, S. T. Nguyen and J. T. Hupp, J. Am. Chem. Soc., 2011, 133, 15858–15861 CrossRef CAS PubMed
- Z. Zhang, L. Zhang, L. Wojtas, P. Nugent, M. Eddaoudi and M. J. Zaworotko, J. Am. Chem. Soc., 2011, 134, 924–927 CrossRef PubMed
- M.-H. Xie, X.-L. Yang, C. Zou and C.-D. Wu, Inorg. Chem., 2011, 50, 5318–5320 CrossRef CAS PubMed
- J. A. Johnson, X. Zhang, T. C. Reeson, Y.-S. Chen and J. Zhang, J. Am. Chem. Soc., 2014, 136, 15881–15884 CrossRef CAS PubMed
- J. A. Johnson, J. Luo, X. Zhang, Y.-S. Chen, M. D. Morton, E. Echeverría, F. E. Torres and J. Zhang, ACS Catal., 2015, 5, 5283–5291 CrossRef CAS
- Y. Liu, D. Chen, X. Li, Z. Yu, Q. Xia, D. Liang and H. Xing, Green Chem., 2016, 18, 1475–1481 RSC
- L. Huang, J. Zhao, S. Guo, C. Zhang and J. Ma, J. Org. Chem., 2013, 78, 5627–5637 CrossRef CAS PubMed
- G. N. Papadopoulos and C. G. Kokotos, Chem. - Eur. J., 2016, 22, 6964–6967 CrossRef CAS PubMed
- E. Arceo, E. Montroni and P. Melchiorre, Angew. Chem., Int. Ed., 2014, 53, 12064–12068 CrossRef CAS PubMed
- Q.-Y. Li, Z. Ma, W.-Q. Zhang, J.-L. Xu, W. Wei, H. Lu, X. Zhao and X.-J. Wang, Chem. Commun., 2016, 52, 11284–11287 RSC
- P. Wu, C. He, J. Wang, X. Peng, X. Li, Y. An and C. Duan, J. Am. Chem. Soc., 2012, 134, 14991–14999 CrossRef CAS PubMed
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed
- L. Zeng, T. Liu, C. He, D. Shi, F. Zhang and C. Duan, J. Am. Chem. Soc., 2016, 138, 3958–3961 CrossRef CAS PubMed
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed
- R. W. Larsen and L. Wojtas, J. Mater. Chem. A, 2013, 1, 14133–14139 CAS
- S. Han, Y. Wei and B. A. Grzybowski, Chem. - Eur. J., 2013, 19, 11194–11198 CrossRef CAS PubMed
- X. Wang, W. Lu, Z.-Y. Gu, Z. Wei and H.-C. Zhou, Chem. Commun., 2016, 52, 1926–1929 RSC
- H. N. Miras, L. Vilà-Nadal and L. Cronin, Chem. Soc. Rev., 2014, 43, 5679–5699 RSC
- A. Hiskia, A. Mylonas and E. Papaconstantinou, Chem. Soc. Rev., 2001, 30, 62–69 RSC
- J. Song, Z. Luo, D. K. Britt, H. Furukawa, O. M. Yaghi, K. I. Hardcastle and C. L. Hill, J. Am. Chem. Soc., 2011, 133, 16839–16846 CrossRef CAS PubMed
- C.-Y. Sun, S.-X. Liu, D.-D. Liang, K.-Z. Shao, Y.-H. Ren and Z.-M. Su, J. Am. Chem. Soc., 2009, 131, 1883–1888 CrossRef CAS PubMed
- Z.-M. Zhang, T. Zhang, C. Wang, Z. Lin, L.-S. Long and W. Lin, J. Am. Chem. Soc., 2015, 137, 3197–3200 CrossRef CAS PubMed
- X. J. Kong, Z. Lin, Z. M. Zhang, T. Zhang and W. Lin, Angew. Chem., 2016, 128, 6521–6526 CrossRef
- R. Liang, R. Chen, F. Jing, N. Qin and L. Wu, Dalton Trans., 2015, 44, 18227–18236 RSC
- D. Shi, C. He, W. Sun, Z. Ming, C. Meng and C. Duan, Chem. Commun., 2016, 52, 4714–4717 RSC
- K. Watanabe, D. Menzel, N. Nilius and H.-J. Freund, Chem. Rev., 2006, 106, 4301–4320 CrossRef CAS PubMed
- P. V. Kamat, J. Phys. Chem. B, 2002, 106, 7729–7744 CrossRef CAS
- P. Wang, B. Huang, Y. Dai and M.-H. Whangbo, Phys. Chem. Chem. Phys., 2012, 14, 9813–9825 RSC
- H. R. Moon, D.-W. Lim and M. P. Suh, Chem. Soc. Rev., 2013, 42, 1807–1824 RSC
- A. Aijaz and Q. Xu, J. Phys. Chem. Lett., 2014, 5, 1400–1411 CrossRef CAS PubMed
- Q.-L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468–5512 RSC
- D. Wang and Z. Li, J. Catal., 2016, 342, 151–157 CrossRef CAS
- D. Sun and Z. Li, J. Phys. Chem. C, 2016, 120, 19744–19750 CAS
- L. Chen, Y. Peng, H. Wang, Z. Gu and C. Duan, Chem. Commun., 2014, 50, 8651–8654 RSC
- Q. Yang, Q. Xu, S. H. Yu and H. L. Jiang, Angew. Chem., 2016, 128, 3749–3753 CrossRef
- T. Toyao, M. Saito, Y. Horiuchi, K. Mochizuki, M. Iwata, H. Higashimura and M. Matsuoka, Catal. Sci. Technol., 2013, 3, 2092–2097 CAS
- L. Shen, M. Luo, L. Huang, P. Feng and L. Wu, Inorg. Chem., 2015, 54, 1191–1193 CrossRef CAS PubMed
- A. L. Linsebigler, G. Lu and J. T. Yates Jr, Chem. Rev., 1995, 95, 735–758 CrossRef CAS
- A. Mills and S. Le Hunte, J. Photochem. Photobiol., A, 1997, 108, 1–35 CrossRef CAS
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS
- L. Shen, S. Liang, W. Wu, R. Liang and L. Wu, J. Mater. Chem. A, 2013, 1, 11473–11482 CAS
- F. Ke, L. Wang and J. Zhu, Nano Res., 2015, 8, 1834–1846 CrossRef CAS
- S. Abedi and A. Morsali, ACS Catal., 2014, 4, 1398–1403 CrossRef CAS
- J. Gao, J. Miao, P.-Z. Li, W. Y. Teng, L. Yang, Y. Zhao, B. Liu and Q. Zhang, Chem. Commun., 2014, 50, 3786–3788 RSC
- A. Fateeva, P. A. Chater, C. P. Ireland, A. A. Tahir, Y. Z. Khimyak, P. V. Wiper, J. R. Darwent and M. J. Rosseinsky, Angew. Chem., 2012, 124, 7558–7562 CrossRef
| This journal is © The Royal Society of Chemistry 2017 |