
Using water adsorption measurements to access the chemistry of defects in the metal–organic framework UiO-66†
Stefano
Dissegna
a,
Rifan
Hardian
b,
Konstantin
Epp
a,
Gregor
Kieslich
 a,
Marie-Vanessa
Coulet
b,
Philip
Llewellyn
b and
Roland A.
Fischer
*a
a,
Marie-Vanessa
Coulet
b,
Philip
Llewellyn
b and
Roland A.
Fischer
*a
aChair of Inorganic and Metal-Organic Chemistry, Faculty of Chemistry and Catalysis Research Center, Technical University of Munich, Lichtenbergstraße 4, 85748 Garching, Germany. E-mail: roland.fischer@tum.de; Fax: +49 (0)89 289 13194; Tel: +49 (0)89 289 13081
bAix-Marseille Univ, CNRS, MADIREL (UMR 7246), Centre de St. Jérôme, 13397 Marseille cedex 20, France
First published on 23rd February 2017
Abstract
Tailoring defects in metal–organic frameworks is important for enhancing sorption and reaction properties. Defects in UiO-66 have been characterized for the first time by using water adsorption measurements. We found that the defect-induced hydrophilicity, quantitatively expressed by the Henry constant and the saturation water uptake, correlates well with the catalytic performance in the cyanosilylation of benzaldehyde.
Metal–organic frameworks (MOFs) are a fascinating class of materials that offer ample opportunities in the research areas of catalysis, gas adsorption, gas separation and related fields.1–4 MOFs are porous materials composed of interconnecting inorganic and organic moieties,5,6 which can be tuned to meet certain requirements.7 For instance, by varying the organic linker, many different structures with tuneable inner pore volumes, e.g. micro or mesoporosity, can be obtained.8 Initially, MOFs have gained a lot of attention in the context of gas-storage applications; however, their thermal and chemical stability have limited possible (industrial) applications so far.9,10 A step forward in this context has been made with Zr-based MOFs, e.g. UiO-66 and PCN-222,11–13 which are stable in most chemical solvents with decomposition temperatures as high as 400 °C.14
When dealing with crystalline materials, defects and disorder are steady companions at temperatures above 0 K. Recently, it has been recognised that defects can be used to alter the properties of MOFs, especially their catalytic activity and sorption behaviour.15–20 For instance, (defective) Zr-based MOFs have been used as Lewis acid catalysts in various reactions, including Meerwein reduction,21 esterification,22 hydrogenation of aromatic compounds,23 epoxide ring-opening reaction and others.19 Despite the large interest of the community in this field, there is a current debate on what is the most effective set of experiments to determine the nature of defects, e.g. concentration, type of defects and their chemistry.
Our work follows on from the important findings of Vermoortele21 and Wu,24 who used different modulators, e.g. acetic acid (AA) and trifluoroacetic acid (TFA) for the incorporation of defects in UiO-66. Here, we use varying amounts of AA and TFA as modulators to prepare UiO-66 samples with an increasing amount of defects.21,24,25 Subsequently, water sorption measurements were performed to elucidate the chemistry of the incorporated defects, in particular their hydrophilicity which can be directly linked to their Lewis acidity. Additionally, we use the cyanosilylation of benzaldehyde as a catalytic test reaction to probe the Lewis acidity of the defects in UiO-66 directly.26,27 Synthetically, 16, 33, 67 and 100 equivalents of AA with respect of zirconia were used to prepare defective UiO-66 and samples are denoted as 16AA, 33AA, 67AA, 100AA, respectively. For TFA as a modulator, 1 and 10 equivalents were used and the samples are denoted as 1TFA and 10TFA (for the proposed sum formulas see Table S2†). Unmodulated UiO-66 (UiO-66 ref) was also synthesized as a reference sample, following a modified procedure reported by Shearer et al.28 The details of the synthesis conditions can be found in the ESI.†
After synthesis, the phase purity of all the samples was confirmed using powder X-ray diffraction (see ESI† Fig. S1).12,28 Additionally, we like to note that no intensity in the low theta region (2–6θ°) was observed which would indicate the presence of correlated defects in UiO-66 as observed by Goodwin and co-workers.29 Moreover, thermogravimetric analysis (TGA) was performed for all samples, and the results were compared to that of UiO-66 ref to investigate the effect of the incorporated amount of the modulator and the thermal stability (see ESI† Fig. S3). For samples with AA as the modulator, a typical weight loss at approximately 300 °C is observed which corresponds to the dehydroxylation of Zr clusters. At approximately 500 °C, a second weight loss is observed, which was previously assigned to linker decomposition in pure UiO-66.24 In contrast to that, the defective UiO-66 samples prepared with TFA as the modulator exhibit a multistep curve in the TGA. The first weight loss at 320 °C was ascribed earlier to TFA anions directly adsorbed on the coordinatively unsaturated sites (CUS) of the zirconium cluster as well as the dehydroxylation of the Zr cluster.21 The first step qualitatively correlates with the amount TFA used in the synthesis. The thermal stability of these samples is approximately 50 °C lower, as compared to the reference. N2 physisorption measurements were also performed (see Fig. S2 in the ESI†), showing type I isotherms, which is typical for microporous MOFs such as UiO-66. Importantly, the porosity of the samples significantly increases with increasing amount of the modulator (see Table 1). This is in strong agreement with previous results, where the increased porosity has been ascribed to an increasing amount of defects incorporated in the UiO-66 framework.12,24,29–34 For materials prepared with increasing quantities of the AA modulator, several trends can be observed as shown in Table 1 and graphically in the ESI† (Fig. S4–S6). When TFA is used, the nitrogen isotherm data shows that the addition of either small or large amounts of modulator (1 and 10 mol equivalent) leads to an increase of the BET surface area and pore volume. This suggests that TFA creates more defective samples than AA (Fig. 1).
| N2 at 77 K | H2O at 298 K | |||||||
|---|---|---|---|---|---|---|---|---|
| Sample | Modulator (eq.) | BET (m2 g−1) | PSD (nm) | Pore volumea (cm3 g−1) | Pore volumeb (cm3 g−1) | Shape of water ads isotherm | Henry constantc (mol−1 g Pa−1) | Max water ads capacity (cm3 g−1) |
| a Pore volume determined with N2 adsorption. b Pore volume determined with water adsorption, PSD = pore size distribution. c A Henry constant value in the order of 10−7 corresponds to very hydrophobic materials, while values in the order of 10−3 correspond to very hydrophilic materials. For further information please see ref. 30 where other Henry constant values for different types of MOFs are reported. | ||||||||
| UiO-66 ref | 0 | 1068 | 1.16 | 0.37 | 0.35 | 2 steps | 2.04 × 10−5 | 495 |
| 16AA | 16AA | 1131 | 1.2 | 0.38 | 0.37 | 1 step | 1.52 × 10−5 | 519 |
| 33AA | 33AA | 1191 | 1.2 | 0.41 | 0.40 | 1 step | 2.49 × 10−5 | 556 |
| 67AA | 67AA | 1314 | 1.2 | 0.47 | 0.44 | 1 step | 2.27 × 10−4 | 603 |
| 100AA | 100AA | 1334 | 1.2 | 0.50 | 0.48 | 1 step | 7.58 × 10−4 | 650 |
| 1TFA | 1TFA | 1100 | 1.2 & 1.5 | 0.40 | 0.39 | 2 steps | 3.52 × 10−4 | 528 |
| 10TFA | 10 TFA | 1567 | 1.2 & 1.5 | 0.59 | 0.55 | 2 steps | 7.12 × 10−4 | 740 |
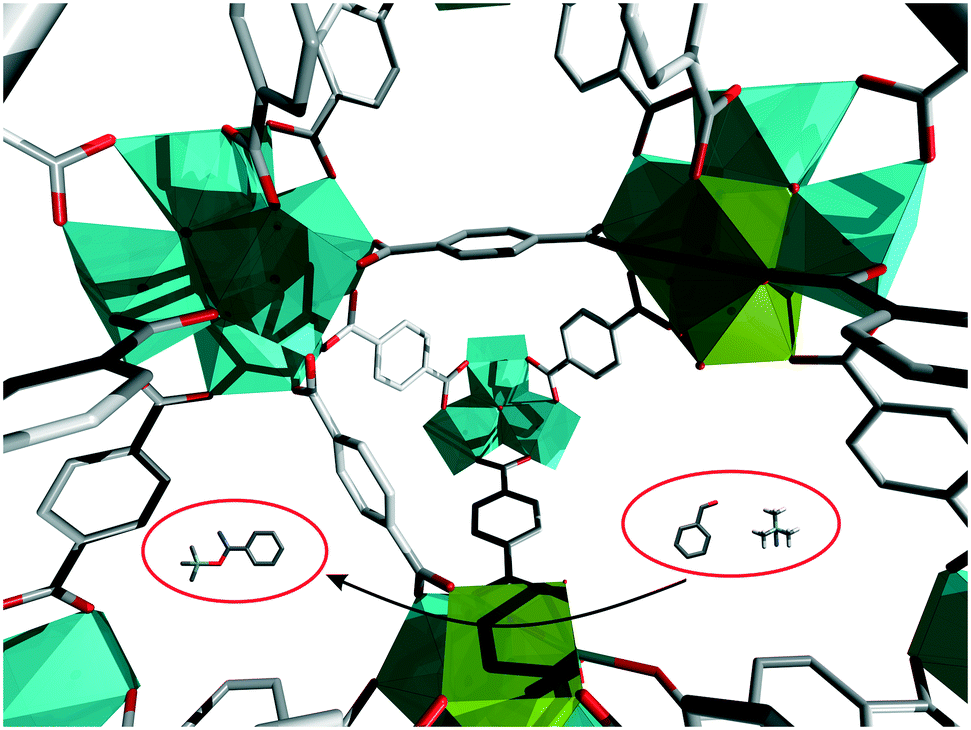 | ||
| Fig. 1 Illustration of the catalytic cyanosilylation of benzaldehyde on a Lewis acidic missing-linker defect in UIO-66. Green polyhedra: clusters with missing linker defects; light blue polyhedra: ideal Zr6 clusters. | ||
In order to access the chemistry of the defects, in particular their hydrophilicity, water adsorption measurements were performed on all the samples in this study. The water adsorption capacity can be determined as the water uptake at partial pressure close to the bulk saturation vapor pressure, p/po; ∼ 1, while the Henry constant, KH, is a quantitative indicator of hydrophilicity. KH corresponds to the slope of the water adsorption isotherm at very low coverage (see ESI† Fig. S7).35 However, this approach is valid assuming an ideal adsorption system. For a more realistic adsorption system (non-ideal gas), an extrapolation of the virial plot of ln(n/p) versus n can be adopted to obtain KH, where n is the mol of adsorbed molecules and p is pressure at low coverage (see ESI† Fig. S8).35
The water adsorption isotherms of the UiO-66 ref and the AA modulated UiO-66 are presented in Fig. 2a. The parameters calculated from these isotherms obtained at 298 K are tabulated in Table 1. It is worth nothing that the Gurvitch rule35 is satisfied as the pore volume obtained with nitrogen at 77 K is comparable with that obtained with water at 298 K. However, slightly lower values of the pore volume are systematically obtained by water sorption which may be explained by the presence of hydrophobic domains within the pores that may repel H2O molecules, thus avoiding complete pore-filling.36 The very low pressure region of the isotherms (Fig. 2a and b) were used to determine the Henry's constant. Interestingly, as can be seen from Table 1, this constant increases with increasing amount of AA. This suggests that AA makes a more hydrophilic surface, because of the missing linker defects which allow the metal site to be more accessible. Furthermore, simulated water sorption isotherms of UiO-66 with missing linker defects indicate an increased hydrophilicity, which is in agreement with our experimental work.31 Interestingly, two steps are observed in the second part of the water adsorption isotherm (range of partial pressure 0.25 to 0.4) for UiO-66 ref. This finding is in agreement with the work of Canivet et al.; however, other studies did not observe a similar feature.30,32,33,37,38 Nevertheless, the observation of this step-like behaviour seems to be highly dependent on the number of data points obtained during water adsorption measurement in the appropriate range of p/po = 0.2–0.4. In earlier works, this behaviour was linked to the dual pore size distribution in UiO-66, and thus is characteristic of the pore filling of the tetrahedral (0.7–0.8 nm, first step) and octahedral (1.1–1.2 nm, second step) cavities respectively.36 In our UiO-66 ref sample, two pore size distributions (PSD) are confirmed by water adsorption but not by N2 adsorption measurements due to the insufficient data points at the very low p/po region. Nevertheless, the addition of AA leads to the disappearance of the first step, suggesting that AA preferentially attacks the smaller pores and enlarges them. Thus, a narrower pore size distribution with only a PSD comparable to that of the octahedral pores can be found for the modulated samples. Furthermore, in the last part of the water sorption isotherm (above p/po = 0.8), the maximum water uptake can be observed, and thus the total pore volume can be calculated. These values also increase as the modulator concentration is increased (Table 1). This can be explained by the additional pore space generated by the defects.32
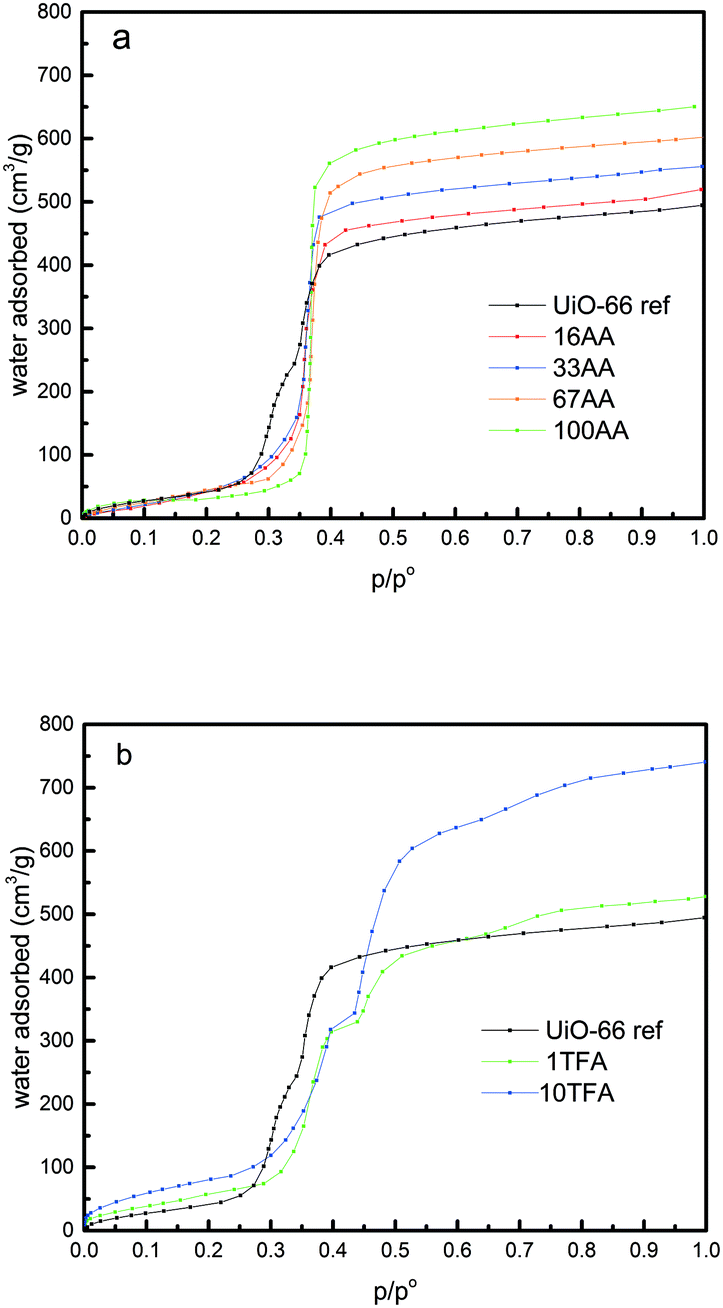 | ||
| Fig. 2 (a) Water sorption isotherms of UiO-66 ref and AA modulated UiO-66, and (b) water sorption isotherms of UiO66 ref and TFA-modulated UiO-66. | ||
Similar to AA, in the TFA case, the Gurvitch rule35 is verified with the pore volume obtained with water at 298 K comparable to the one obtained with nitrogen at 77 K. Similarly, the Henry's constants were evaluated for the TFA samples, and we found that the TFA samples are more hydrophilic than the UiO-66 ref and the AA modulated samples. Interestingly, the shapes of the isotherms are more complex for the TFA-modulated samples. Indeed, in the range of p/po 0.3–0.5 for both TFA samples, two steps can be clearly observed. This behaviour seems to be more prominent than in the UiO-66 ref sample. This may indicate that TFA does not seem to attack preferentially the smaller pores but rather enlarges both cavities. Another interesting observation from the water adsorption experiment on the TFA modulated UiO-66 is the onset of a third step around p/po ∼ 0.8. Since we did not observe this step in UiO-66 ref and in the AA modulated UiO-66, we can propose two interpretations of this effect. (i) This step is related to the creation of a third pore size distribution that results from the merging of some of the two other micropores, or (ii) this is linked to the outer surface or interparticle condensation.36 To propose a possible mechanism, it is worth noting that the weakest point in the UiO-66 structure is the bond between the benzene rings and the terminal carboxyl group in the BDC linker.12 In the case of the AA modulator, the BDC linker is replaced by AA, whereas in the case of TFA modulated UiO-66, the replacement of BDC by TFA is followed by the removal of TFA upon activation.21 This may create larger cavities and more hydrophilic unsaturated zirconium metal sites. A more detailed comparison of the water isotherms of UiO-66 ref, AA-modulated UiO-66, and TFA-modulated UiO-66 is given in Fig. S7 (see the ESI†). Another possible explanation for the water adsorption isotherm depicted in Fig. S7† is it might be due to the nature of the zirconium metal site that could undergo reversible structural rearrangement upon activation and introduction of water vapor.12,33 In the case of UiO-66 ref, it is known that the as-synthesized UiO-66 can readily contain some missing linkers,39,40 and the zirconium metal cluster might exist in the hydroxylated (VIII) and dehydroxylated (VII) forms, probably due to the incomplete activation process. This coexistence could lead to the reversible structural rearrangement upon water adsorption and the readily available missing linkers were terminated by water molecules during water adsorption processes. This termination process, thus, could induce a step in the isotherm. In the case of AA-modulated UiO-66, there is no step observed in the middle range of the partial pressure of water adsorption. This might be linked to the addition of AA that does not only replace some of the BDC linkers but also terminate the readily available missing linkers. Therefore, there is no water-induced missing linker termination during water adsorption and in turn, no step is observed in the isotherm. In the case of TFA-modulated UiO-66, the replacement of some BDC linkers by TFA is followed by the removal of TFA upon activation.21 This creates a bigger cavity and the zirconium metal sites are more exposed to water. In this case, structural rearrangement is more likely to happen and water-induced missing linker termination is required to maintain the charge balance in the defective structure. Therefore, the step in the water adsorption isotherm is more pronounced, representing combination effects between the presence of two pore sizes (as evidenced from the nitrogen adsorption measurement) and the reversible structural changes upon defect termination by water molecules.
Lastly, in order to check if the created defects produce a material with more Lewis acid sites, we chose the cyanosilylation of benzaldehyde as a test reaction, which requires the presence of such Lewis acid sites (Scheme 1).
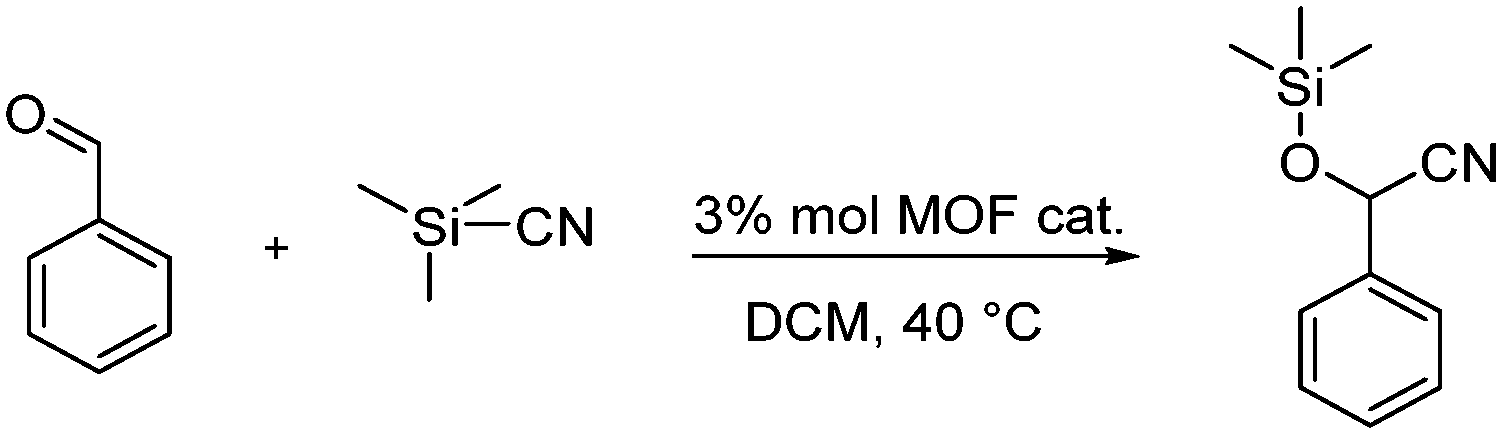 | ||
| Scheme 1 Cyanosilylation of benzaldehyde with trimethylsilylcyanide (TMSCN) in CH2Cl2 (DCM) to give the corresponding cyanohydrin under the Lewis-acidic Zr-MOF. | ||
In accordance with our expectation, the defect engineered TFA-modulated samples reveal a significantly higher catalytic activity when compared to their “ideal” UiO-66 counterpart, which is considered as defect-free. Notably, a slight induction period is observed at the beginning of the reaction, which can be attributed to the reversible formation of a benzoin-condensation product (Fig. 3).
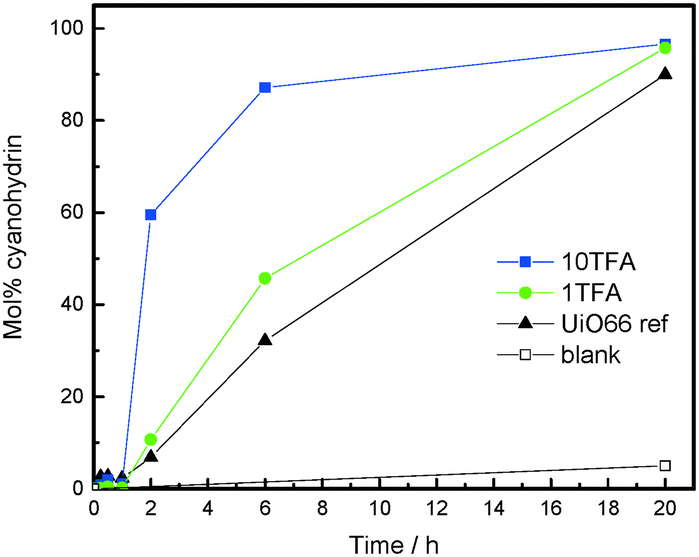 | ||
| Fig. 3 Time–yield plot of the cyanohydrin produced by the cyanosilylation of benzaldehyde with trimethylsilylcyanide (TMSCN) catalyzed over 10TFA (blue), 1TFA (green) and UiO66 ref (black) samples in comparison with the blank sample (black, open), in which no catalyst was used. | ||
Moreover, the catalytic activity nicely correlates with the degree of TFA modulation, whereby the created defects and the lowered coordination number of the Zr6 clusters cause an improvement in the reaction rate. However, it is unclear if the drastic impact on the catalytic activity is only caused by the defect sites and/or by the increased surface area of the TFA-modulated samples which should also help to overcome diffusion limitations. Most likely, both phenomena contribute to the boost in the catalytic performance. Furthermore, all the tested catalysts show excellent stability to the applied reaction conditions and could be recycled for at least two consecutive catalytic runs, which is confirmed by PXRD (Fig. S9 and Table S1 in the ESI†). In order to investigate the heterogeneity of the underlying reaction, a hot-filtration test was conducted which revealed that the reaction was cancelled after the catalyst was separated from the reaction solution (Fig. S10 ESI†).
Conclusions
Herein, we report water adsorption measurements as a key tool to study the generated defects in AA and TFA modulated UiO-66 samples. We found that increasing the amount of the modulator causes an increase in the surface area (BET), total water uptake and hydrophilicity, especially in the TFA modulated samples with respect to unmodulated UiO-66 sample. By this modulation approach, it is possible to increase the hydrophilicity, which is reflected by the Henry constant, of UiO-66 by one order of magnitude. Furthermore, we want to highlight that it is possible to link the hydrophilicity and the catalytic activity. Therefore, we chose the cyanosilylation of benzaldehyde as a test reaction, which exploited the Lewis acid sites generated by missing linker defects. Hereby, we found that increasing the amount of TFA has a beneficial effect on the catalytic activity. In conclusion, we would like to introduce water adsorption measurement as a complementary characterization method in order to access the chemistry of defects. Compared to other techniques, it offers a direct way to measure the hydrophilicity, which can be used to predict the catalytic properties for Lewis acid-based reactions.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No. 641887 (project acronym: DEFNET). GK acknowledges the support from the Fonds der chemischen Industrie (Liebig fellowship).Notes and references
- A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed
.
- C. Rösler and R. A. Fischer, CrystEngComm, 2014, 17, 199–217 RSC
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed
- M. A. Nasalevich, M. van der Veen, F. Kapteijn and J. Gascon, CrystEngComm, 2014, 16, 4919 RSC
- M. Vandichel, J. Hajek, F. Vermoortele, M. Waroquier, D. E. De Vos and V. Van Speybroeck, CrystEngComm, 2015, 17, 395–406 RSC
- O. M. Yaghi, M. O’Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC
- M. Bosch, M. Zhang, H.-C. Zhou, M. Bosch, M. Zhang and H.-C. Zhou, Adv. Chem., 2014, 2014, 1–8 CrossRef
- Z. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54, 7234–7254 CrossRef CAS PubMed
- D. Feng, Z. Y. Gu, J. R. Li, H. L. Jiang, Z. Wei and H. C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS PubMed
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS
- J. B. DeCoste, G. W. Peterson, H. Jasuja, T. G. Glover, Y. Huang and K. S. Walton, J. Mater. Chem. A, 2013, 1, 5642 CAS
- Z. Fang, J. P. Dürholt, M. Kauer, W. Zhang, C. Lochenie, B. Jee, B. Albada, N. Metzler-Nolte, A. Pöppl, B. Weber, M. Muhler, Y. Wang, R. Schmid and R. A. Fischer, J. Am. Chem. Soc., 2014, 136, 9627–9636 CrossRef CAS PubMed
- F. Vermoortele, M. Vandichel, B. Van De Voorde, R. Ameloot, M. Waroquier, V. Van Speybroeck and D. E. De Vos, Angew. Chem., Int. Ed., 2012, 51, 4887–4890 CrossRef CAS PubMed
- J. Canivet, M. Vandichel and D. Farrusseng, Dalton Trans., 2016, 4090–4099 RSC
- A. W. Thornton, R. Babarao, A. Jain, F. Trousselet and F.-X. Coudert, Dalton Trans., 2016, 45, 4352–4359 RSC
- Y. Liu, R. C. Klet, J. T. Hupp and O. Farha, Chem. Commun., 2016, 52, 7806–7809 RSC
- O. Kozachuk, I. Luz, F. X. Llabrés i Xamena, H. Noei, M. Kauer, H. B. Albada, E. D. Bloch, B. Marler, Y. Wang, M. Muhler and R. A. Fischer, Angew. Chem., Int. Ed., 2014, 53, 7058–7062 CrossRef CAS PubMed
- F. Vermoortele, B. Bueken, G. Le Bars, B. Van De Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. Van Speybroeck, C. Kirschhock and D. E. De Vos, J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed
- F. G. Cirujano, A. Corma and F. X. Llabrés i Xamena, Chem. Eng. Sci., 2015, 124, 52–60 CrossRef CAS
- A. M. Rasero-Almansa, A. Corma, M. Iglesias and F. Sánchez, Green Chem., 2014, 16, 3522–3527 RSC
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, J. Am. Chem. Soc., 2013, 135, 10525–10532 CrossRef CAS PubMed
- G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28, 3749–3761 CrossRef
- F.-G. Xi, Y. Yang, H. Liu, H.-F. Yao and E.-Q. Gao, RSC Adv., 2015, 5, 79216–79223 RSC
- A. Dhakshinamoorthy, M. Alvaro, A. Corma and H. Garcia, Dalton Trans., 2011, 40, 6344 RSC
- G. C. Shearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Chem. Mater., 2014, 26, 4068–4071 CrossRef CAS
- M. J. Cliffe, W. Wan, X. Zou, P. A. Chater, A. K. Kleppe, M. G. Tucker, H. Wilhelm, N. P. Funnell, F.-X. Coudert and A. L. Goodwin, Nat. Commun., 2014, 5, 4176 CAS
- J. Canivet, J. Bonnefoy, C. Daniel, A. Legrand, B. Coasne and D. Farrusseng, New J. Chem., 2014, 38, 3102 RSC
- P. Ghosh, Y. J. Coló and R. Q. Snurr, Chem. Commun., 2014, 50, 11329–11331 RSC
- H. Furukawa, F. Gadara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed
- A. D. Wiersum, E. Soubeyrand-Lenoir, Q. Yang, B. Moulin, V. Guillerm, M. Ben Yahia, S. Bourrelly, A. Vimont, S. Miller, C. Vagner, M. Daturi, G. Clet, C. Serre, G. Maurin and P. L. Llewellyn, Chem. – Asian J., 2011, 6, 3270–3280 CrossRef CAS PubMed
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS
-
F. Rouquerol, J. Rouquerol, K. S. W. Sing, P. Llewellyn and G. Maurin, Adsorption by powders and porous solids: Principles, Methodology and Applications, Elsevier Ltd, 2014 Search PubMed
- F. Jeremias, V. Lozan, S. K. Henninger and C. Janiak, Dalton Trans., 2013, 42, 15967 RSC
- H. Jasuja and K. S. Walton, J. Phys. Chem. C, 2013, 117, 7062–7068 CAS
- P. M. Schoenecker, C. G. Carson, H. Jasuja, C. J. J. Flemming and K. S. Walton, Ind. Eng. Chem. Res., 2012, 51, 6513–6519 CrossRef CAS
- S. Ling and B. Slater, Chem. Sci., 2016, 7, 4706–4712 RSC
- C. A. Trickett, K. J. Gagnon, S. Lee, F. Gándara, H. B. Bürgi and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 11162–11167 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ce00224f |
| This journal is © The Royal Society of Chemistry 2017 |