
Two lanthanide-based metal–organic frameworks for highly efficient adsorption and removal of fluoride ions from water†
Aiqing
Ma‡
a,
Fei
Ke‡
*b,
Jing
Jiang
b,
Qiaoyu
Yuan
b,
Zhidong
Luo
a,
Jianqiang
Liu
 *a and
Abhinav
Kumar
c
*a and
Abhinav
Kumar
c
aSchool of Pharmacy and Guangdong Key Laboratory for Research and Development of Natural Drugs, Guangdong Medical University, Dongguan, 523808, P. R. China. E-mail: jianqiangliu8@gdmc.edu.cn
bDepartment of Applied Chemistry and State Key Laboratory of Tea Plant Biology and Utilization, Anhui Agricultural University, Hefei 230036, P.R. China. E-mail: kefei@ahau.edu.cn
cDepartment of Chemistry, Faculty of Science, University of Lucknow, Lucknow 226 007, India
First published on 21st March 2017
Abstract
The contamination of water with fluoride (F−) is a source of mounting concern for global public health, and the removal of fluoride is quite important and challenging. In this study, two new lanthanide-based metal–organic frameworks (MOFs), {[Ce(L1)0.5(NO3)(H2O)2]·2DMF} (1) and [Eu3(L2)2(OH)(DMF)0.22(H2O)5.78]·guest (2) (H4L1 = 2,5-di(3′,5′-dicarboxylphenyl)benzene and H4L2 = 3,5-bis(isophthalic acid)-1H-1,2,4-triazole) are designed, synthesized and characterized. Both MOFs are tested for their adsorption capacity for fluoride ions and different uptake times from contaminated water. The investigation indicates that 1 displays a much higher adsorption capacity (103.95 mg g−1) and faster uptake rates (1.79 g mg−1 min−1) for fluoride ion than that of 2. The presented investigation is the first report in which lanthanide-based MOFs are used for the removal of fluoride ions from water.
Introduction
The enormous industrialization and rapid techno-economic growth in the human civilization have led to environmental pollution, which may lead to the accumulation of toxic elements in living organisms and cause an impending global crisis since these unwanted accumulations induce several biological disorders and diseases.1 Currently, fluoride ion is one of the most abundant contaminants in water. Fluoride, a double-edged sword in drinking water, is one of the essential trace elements for the human body.2 However, excessive uptake of fluoride can lead to serious health problems, such as dental caries and skeletal fluorosis.3 The maximum permissible fluoride concentration level of 1.5 mg L−1 in drinking water has been recommended by the World Health Organization (WHO).4 However, the fluoride concentration has been found to be as high as 30 mg L−1 in more than 25 countries from Africa, Europe and America.5 In this regard, fluoride remediation has been an active field of research for a long time. To date, a variety of technologies and methods have been proposed for the removal of unwarranted fluoride from water, including ion-exchange, chemical precipitation, membrane-based processes, and adsorption.6,7 Owing to its simple operation, effectiveness and low cost, the adsorption method is considered as one of the most promising and applicable approaches for defluoridation.8 Consequently, numerous adsorbents, such as activated alumina,9 zirconium-based sorbents,10 calcium-based sorbents,11 and iron-based sorbents,12 have been used for effective fluoride removal from water. In addition to these inorganic adsorbents, multivalent lanthanide-based rare-earth metal composites have high specific affinity for undergoing complexation with fluoride due to their hard acid nature, for example, Fe–Al–Ce trimestral hydrous oxide,13 Mn–La bimetal composite,14 and La-loaded magnetic cationic hydrogel.15 Although these lanthanide-based inorganic adsorbents possess a positive charge, the surface area of these adsorbents are very low which thereby results in a low fluoride adsorption capacity. Therefore, there is still plenty of scope to explore the unfulfilled potentials of lanthanide-based complexes for the development of high capacity fluoride adsorbents.As a class of newly developed porous materials, metal–organic frameworks (MOFs), which are conventionally designed and constructed using a large number of metal ions/clusters and organic ligands through coordination bonds, are considered as an attractive alternative platform for the adsorptive capture and removal of fluoride-containing species.16 Compared with traditional porous materials, the interest in MOFs-based materials arises because of their extraordinarily high porosities, diverse functional sites, and easy modulation of their pore size, which is achieved by changing the connectivity of the inorganic vertices and the organic braces.17 These distinct characteristics make them very promising for a wide variety of potential applications,18–21 such as catalysis,18 gas storage and separation,19 and drug delivery.20 In terms of the adsorption of hazardous molecules and ions from aqueous solution, MOFs are promising materials for adsorption-related applications because the size, shape and composition of their pores can be controlled for the uptake of specific hazardous molecules.22 Some porous MOFs have been explored for the removal of hazardous materials and they showed excellent adsorption abilities and selectivity.23 However, most reports concerning the adsorption and removal of contaminants from water using MOFs are limited to heavy metal ions, organic dyes, oil droplets, and toxic sulfur-containing compounds.24–27 To date, sporadic pioneering studies have been reported for the removal of fluoride using MOFs as adsorbents (e.g., AlFu MOF,28 UiO-66-NH2,29 MIL-88(Fe),30 and MIL-96(Al)31). However, to the best of our knowledge, there is no report on the utilization of lanthanide-based MOFs as adsorbents to remove fluoride from water.
Considering the abovementioned facts and in the quest of new lanthanide-based MOFs that may act as efficient adsorbents of fluoride two MOFs, {[Ce(L1)0.5(NO3)(H2O)2]·2DMF} (1) and [Eu3(L2)2(OH)(DMF)0.22(H2O)5.78]·guest (2) (H4L1 = 2,5-di(3′,5′-dicarboxylphenyl)benzene and H4L2 = 3,5-bis(isophthalic acid)-1H-1,2,4-triazole) (Scheme S1†), were designed, characterized and synthesized and their possible application as adsorbents for fluoride were explored, and the results of these investigations are presented herein.
Experimental
Materials and methods
All purchased chemicals were used without further purification. Powder X-ray diffraction (PXRD) data was collected on a Bruker D8 ADVANCE X-ray diffractometer with Cu-Kα radiation (λ = 1.5418 Å) at 50 kV, and 20 mA with a scanning rate of 6° min−1 and a step size of 0.02°. Fourier transform infrared (FT-IR) spectra were measured using a Nicolet Impact 750 FTIR in the range of 400–4000 cm−1 and KBr pellets. Thermogravimetric analyses were performed under N2 atmosphere from room temperature to 650 °C at a heating rate of 10 °C min−1, using an SDT Q600 thermogravimetric analyzer. The fluoride ion stock solution (500 ppm) was prepared by dissolving NaF in deionized water, and the test solutions (12.5 to 400 ppm) were made by the subsequent dilution of the fluoride stock solution with deionized water.X-ray crystallography
Single crystal X-ray diffraction data were collected on a Bruker SMART APEX diffractometer equipped with a graphite monochromated MoKα radiation source (λ = 0.71073 Å) using the ω-scan technique. The intensities were corrected for absorption effects using SADABS. The structures were solved by direct methods (SHLEXL-2014)32 and refined by a full-matrix least-squares procedure based on F2. All the hydrogen atoms were generated geometrically and refined isotropically using the riding model. All non-hydrogen atoms were refined with anisotropic displacement parameters. In 1, the free DMF solvent molecules are disordered, and the attempts to locate and refine all the solvent molecule peaks were unsuccessful. Diffused electron densities resulting from these solvent molecules were removed from the data set using the SQUEEZE routine of PLATON and refined further using the data generated. The contents of the removed solvent region are not represented in the unit cell contents in the crystal data. The final formula of 1 from the SQUEEZE results was determined in combination with elemental analysis and thermogravimetric analysis data. The crystallographic details and selected bond dimensions for 1 are listed in Tables S2 and S3.† CCDC: 1518722 for 1.Syntheses of 1–2
Results and discussion
Structural feature
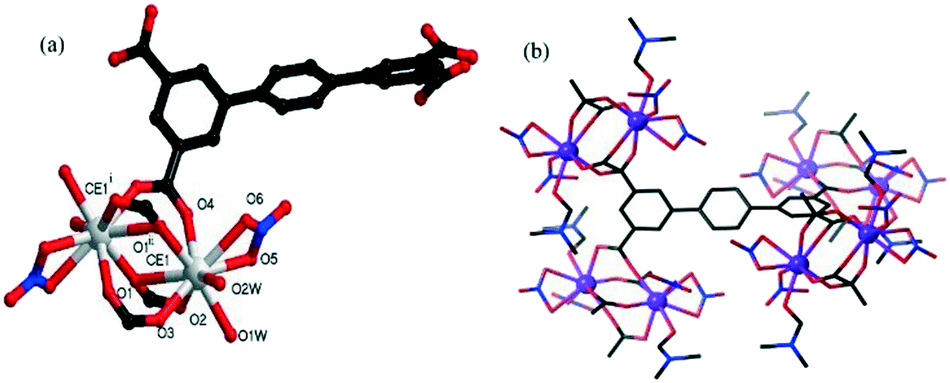 | ||
| Fig. 1 (a) Local ligand and metal coordination geometries in the structure of 1 (symmetric codes: (i) −x, y, −z + 1/2 and (ii) x + 1/2, y + 1/2); and (b) the second building unit for the dinuclear Ce2 clusters. | ||
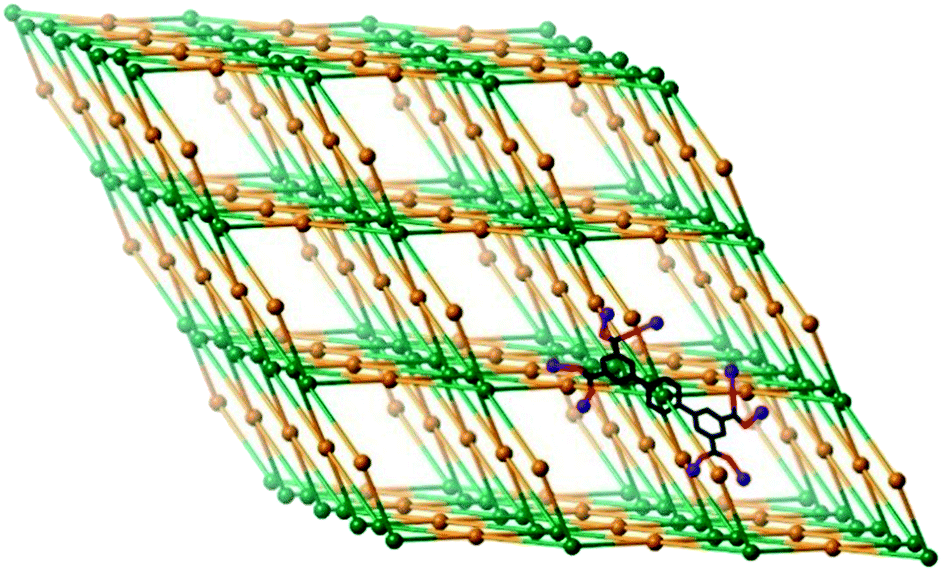 | ||
| Fig. 2 Schematic of the 3D PtS network formed in the structure of 1. Green nodes represent the ligands, whereas the orange nodes represent the Ce2 dinuclear clusters. A single ligand of the unsimplified structure, with coordinated Ce atoms, is shown in the bottom right corner. | ||
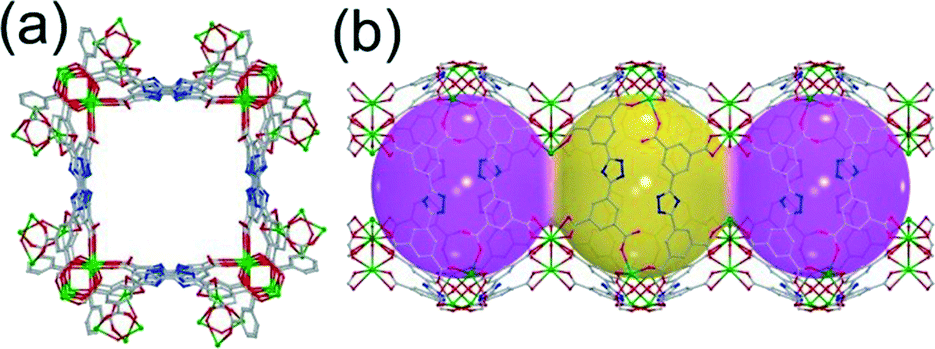 | ||
| Fig. 3 (a) 3D framework viewed along the c axis, and (b) 1D square channels in 2. | ||
Effect of pH on adsorption
Solution pH is one of the most significant factors in the adsorption process because it can influence the surface charge and plays an important role in the adsorption capacity of the adsorbent. To evaluate the influence of solution pH on the adsorption efficiency for fluoride, 30 mg of the lanthanide-based MOFs adsorbents (1 and 2) were suspended in 15 mL fluoride ions solution (12.5 ppm) under constant shaking at various pH (from 2 to 10), and the results are presented in Fig. 4. It can be observed that the adsorption is higher at lower pH (3–7) and drops drastically after pH 8, which can be attributed to the adsorption competition between F− and OH−. At a lower pH, the lanthanide-based MOFs adsorbents are positively charged and this nucleophilic replacement of OH− by F− is very easy, whereas such nucleophilic replacement is not preferred at a higher pH because of the abundance of OH−. At pH 2, however, the MOFs were found to have negligible adsorption efficiency of fluoride, which may be due to the decomposition of the MOFs in such strong acidic conditions. Therefore, it is apparent that the best pH range for the adsorption of fluoride from water is from 3 to 7. However, the pH value of the actual prepared fluoride solution was 7.09, and thus we did not adjust the pH during the subsequent experiment.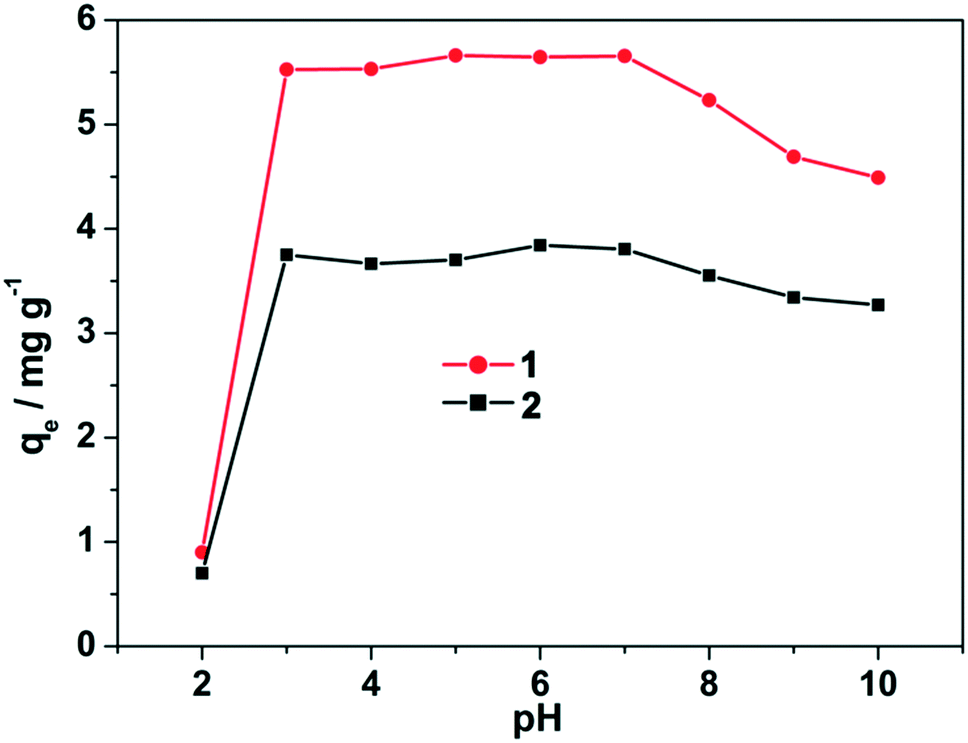 | ||
| Fig. 4 Effects of pH on the removal efficiency of fluoride by the MOF adsorbents at 298 K with the initial fluoride concentration of 12.5 ppm. | ||
Adsorption kinetics for fluoride removal from water
In order to evaluate their fluoride adsorption rates, fluoride was adsorbed on 1–2 for different time intervals up to 120 min. The quantity of adsorbed fluoride is displayed in Fig. 5 when the initial fluoride concentration was 12.5 ppm. As can be seen in Fig. 5, the adsorbed quantity of fluoride is in the order of 2 < 1 during the entire adsorption period. The fluoride removal increases rapidly with contact time. Significantly, the adsorption time needed for the saturation of the adsorbed amount over sample 1 is practically completed in only 5 min, which suggests that 1 possesses a rapid adsorption efficiency for the removal of fluoride. In addition, to verify the F− adsorption capacity on 1 and 2, X-ray photoelectron spectroscopy (XPS) studies were carried out on 1@F− and 2@F−. The peak at 681 and 684 eV in the F1s XPS spectra for 1@F− and 2@F−, respectively, is attributed to F− ions (Fig. S2†). Moreover, the powder X-ray diffraction (PXRD) patterns (Fig. S3†) indicate that the basic frameworks of both MOFs remained intact when 1@F− and 2@F− were formed in the ion solutions. The stabilities of 1@F− and 2@F− were also confirmed by IR spectroscopy after the inclusion (Fig. S4†).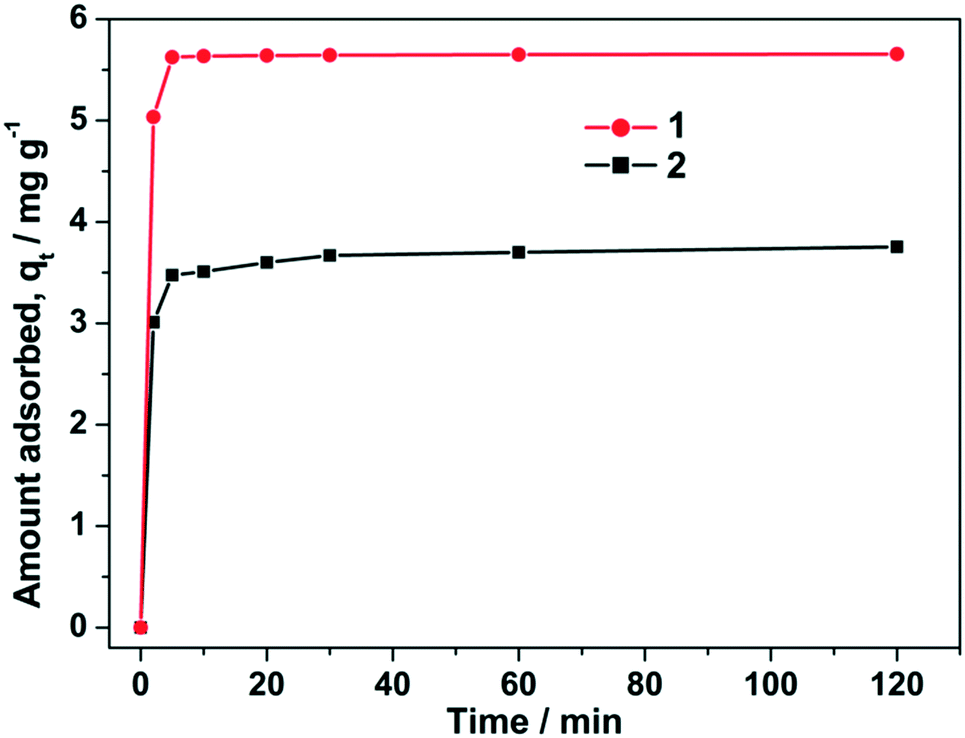 | ||
| Fig. 5 Effect of contact time on the adsorption of fluoride over the two MOFs adsorbents at 298 K with the initial fluoride concentration of 12.5 ppm. | ||
In order to analyze the adsorption kinetics for both MOFs, the kinetics parameters were treated with the pseudo-second-order kinetic equation.34 The final values of k2, qe and R are listed in Table S1.† It has been found that the linear correlation coefficients are very close to 1, which indicates that the adsorption behaviors of fluoride on 1–2 conform with a pseudo-second-order kinetic model. The adsorption kinetic rate constant, k2, for fluoride adsorption is in the order of 2 < 1, which is in good agreement with the adsorbed quantity (Fig. 6). Remarkably, the k2 value for 1 was determined to be 1.79 g mg−1 min−1. This value is superior to many of the previously reported porous adsorbents.29,31,32 Therefore, 1 is the most effective adsorbent for fluoride removal from water in the viewpoint of adsorption amount and rate.
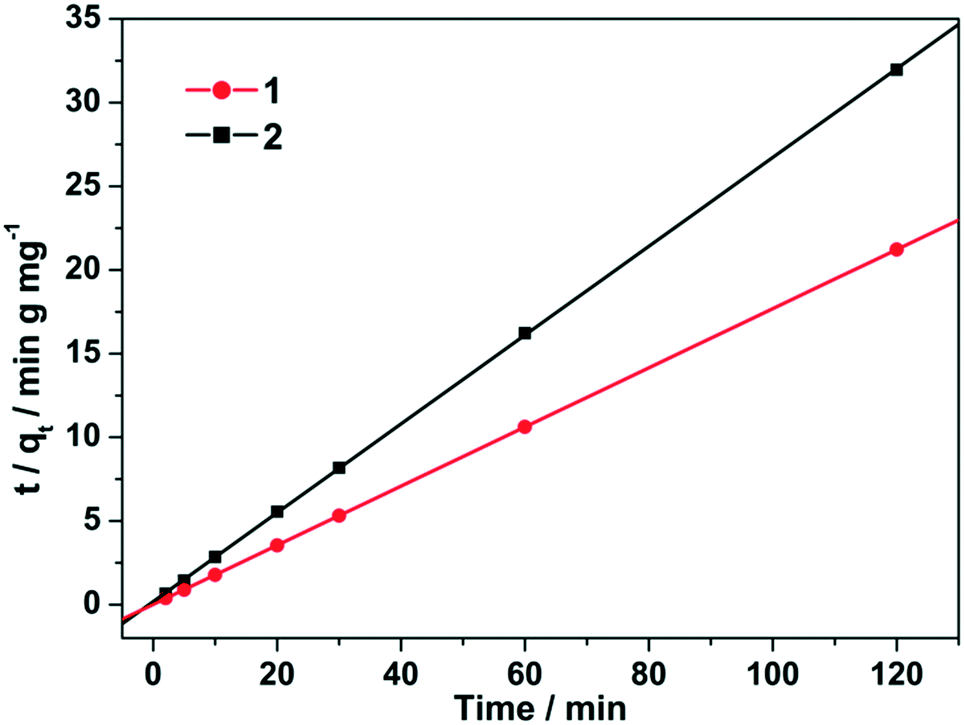 | ||
| Fig. 6 Plots of the pseudo-second-order kinetics of fluoride adsorption over the adsorbents 1 and 2 at 298 K with the initial fluoride concentration of 12.5 ppm. | ||
Adsorption thermodynamics
The removal ability of 1 and 2 was preliminarily tested under a wide range of known fluoride concentrations. The plots of Cevs. qe of the fluoride adsorption at different temperatures are presented in Fig. 7a and b. The adsorption isotherms were obtained after the adsorption of fluoride for a time period of 120 min. The presented data indicates that the adsorption capacities of MOFs 1 and 2 to adsorb fluoride increases with an increase in fluoride equilibrium concentration. Moreover, to quantitatively analyze the adsorption isotherm data, the Langmuir adsorption model was employed to evaluate the maximum adsorption capacity of 1 and 2:35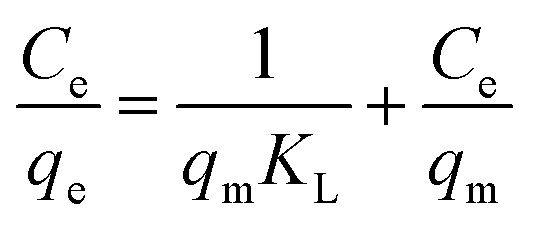
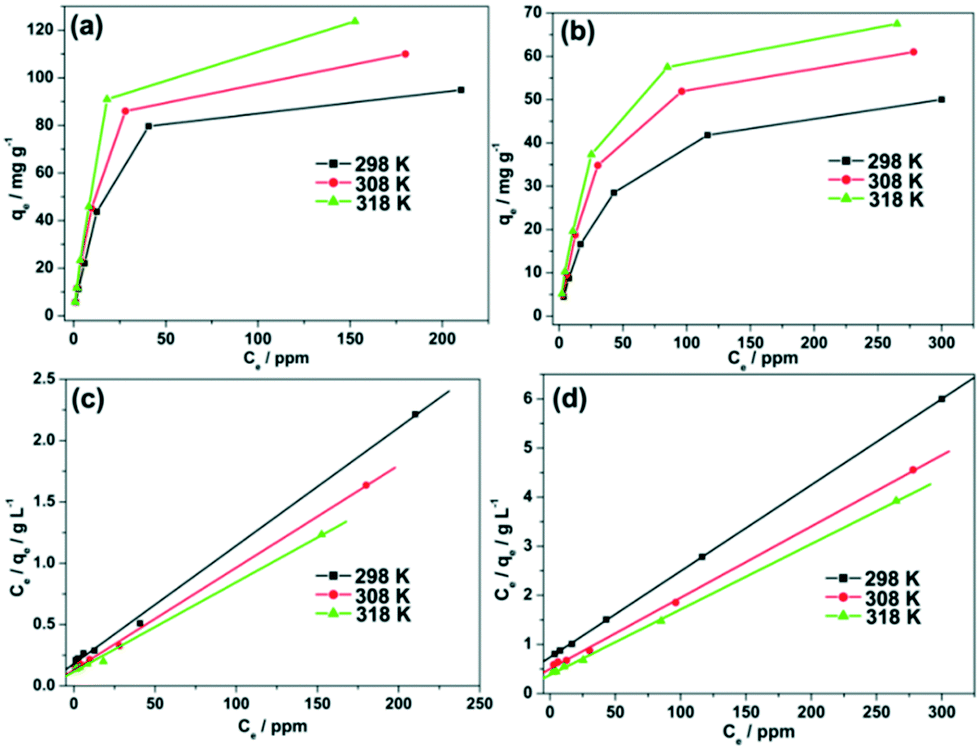 | ||
| Fig. 7 Adsorption isotherms and the linear regression by fitting the equilibrium adsorption data with the Langmuir adsorption model for fluoride adsorption over adsorbents 1 (a and c) and 2 (b and d) at 298 K, 308 K and 318 K. | ||
The plots of Ceversus Ce/qe at different temperatures were well fitted by the linear regression, indicating that the adsorption of fluoride also conforms to the Langmuir adsorption model (Fig. 7c and d).
The maximum adsorption capacity qm for all of the samples is summarized in Table 1. The maximum adsorption capacity of 1 and 2 was calculated to be 103.95 and 57.01 mg g−1 at 298 K, respectively. The coordinatively unsaturated Ce(III) centers in sample 1 were surrounded by –OH and NO3−. The fluoride ion attacked the Ce centers in the frameworks and its adsorption occurred at the Ce center with the simultaneous displacement of –OH and –NO3−, resulting in the formation of Ce–F bonds. The larger adsorption capacity of 1, compared to that of 2, is probably due to the stronger interaction between Ce(III) and the fluoride ions. In comparison to the previously reported adsorption capacities of MIL-96(Al) (31.69 mg g−1)31 and UiO-66-NH2 (58.82 mg g−1),29 the fluoride adsorption capacities presented herein exceed by a factor of ∼3.28 and ∼1.77 for 1 and 2, respectively.
| Adsorbent | Temp. K | q m mg g−1 | ΔG kJ mol−1 | ΔH kJ mol−1 | ΔS J mol−1 K−1 |
|---|---|---|---|---|---|
| 1 | 298 | 103.95 | −4.23 | ||
| 308 | 119.90 | −5.22 | 18.52 | 76.56 | |
| 318 | 137.17 | −5.70 | |||
| 2 | 298 | 57.01 | −0.74 | ||
| 308 | 68.87 | −1.77 | 25.32 | 87.75 | |
| 318 | 75.02 | −2.56 |
The thermodynamic behavior of the adsorption of fluoride on the adsorbents at different temperatures can be obtained from the adsorption free energy (ΔG), enthalpy (ΔH) and entropy (ΔS). The adsorption isotherms and the corresponding Langmuir plots at the temperatures of 298, 308 and 318 K are shown in Fig. 8. Their adsorption capacity will be accompanied by an adsorption temperature (Fig. 7 and Table 1), and thus it can be seen that the adsorption of fluoride on 1 and 2 is endothermic in nature. The change in Gibbs free energy (ΔG) can be calculated using the following equation:35
ΔG = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnK lnK |
The ΔG values of the adsorption of fluoride on 1 and 2 at different temperatures are presented in Table 1. The calculated ΔG values at 298, 308 and 318 K are all negative, which reveals that the adsorption of fluoride on both 1 and 2 are thermodynamically feasible and spontaneous in nature. The ΔG values become more negative when the temperature increases, which suggests that the adsorption process is more favorable at high temperatures. The enthalpy change (ΔH) and entropy change (ΔS) of the adsorption can be calculated:35
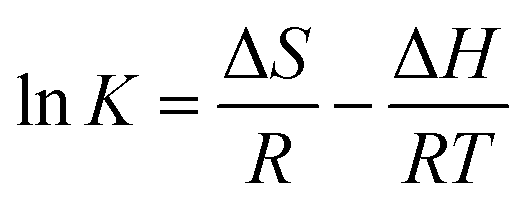
Using above equation, the plot of lnK vs. −1/T is shown in Fig. 8, and the values of ΔH and ΔS can be directly obtained from the slope and intercept of the van't Hoff plot. The fitted values of ΔH are 18.52 and 25.32 kJ mol−1 for 1 and 2, respectively (Table 1). The positive values of ΔH for 1 and 2 also confirm the endothermic nature of the full adsorption process. The endothermic feature of the adsorption process may be ascribed to the stronger interaction between pre-adsorbed water and 1–2. Similar findings were also reported in previous investigations.29
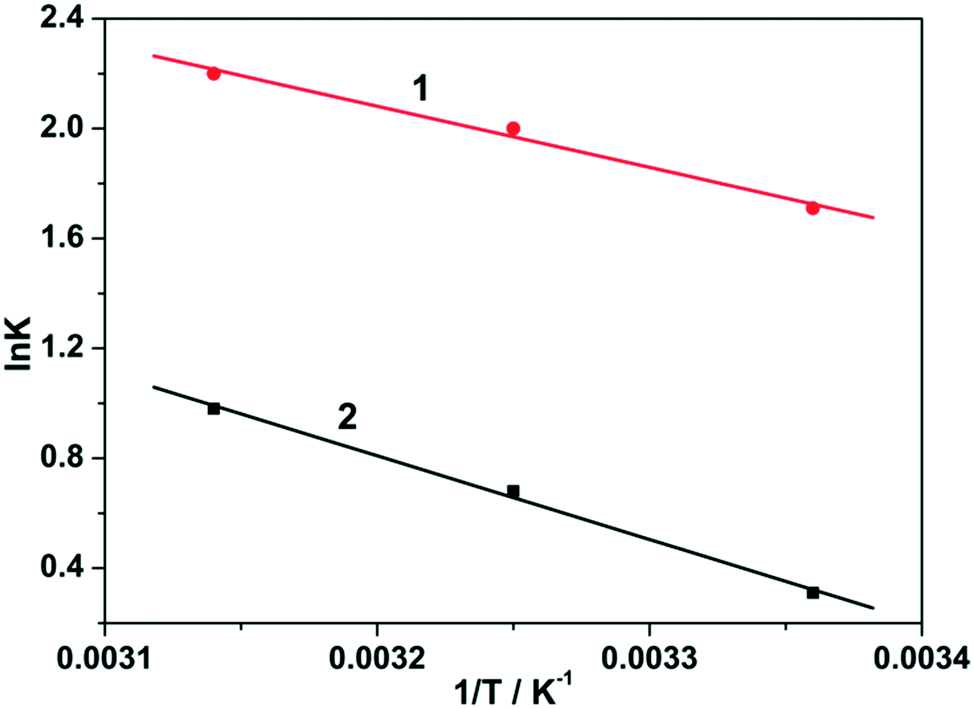 | ||
| Fig. 8 van't Hoff plots of the ΔH and ΔS of the fluoride adsorption over the 1 and 2. | ||
Furthermore, the ΔS parameters obtained from the van't Hoff plots, are 76.56 and 87.75 J mol−1 K−1 for 1 and 2, respectively (Table 1). The positive ΔS values imply an increase in randomness at the solid/solution interface during the adsorption process of fluoride. It is generally known that a large positive ΔS or large negative ΔH will be helpful for spontaneous adsorption (negative ΔG). Herein, both the ΔH and ΔS of the fluoride adsorption over 1 and 2 are positive values. Therefore, the driving force of fluoride adsorption over 1 and 2 in this study is attributed to an entropy effect (ΔS) rather than an enthalpy change (ΔH).
Conclusions
In summary, two lanthanide-based MOFs, 1 and 2, have been used as adsorbents for the removal of fluoride from water, and they show excellent performances. It is found that 1 exhibits a higher adsorption capacity and faster rate of uptake for fluoride than 2. The adsorption capacity of 1 for fluoride was 103.95 mg g−1 at 298 K. The adsorption capacity of fluoride by the lanthanide-based MOFs almost remains constant in the pH range of 3–7. However, the comparison between the adsorption capacities of 1 and 2 suggests that the fluoride adsorption to 1 might involve a strong interaction between fluoride and the Ce site. The kinetics, adsorption isotherm and thermodynamics of fluoride adsorption on 1 and 2 were also investigated in this study. Furthermore, the driving force of fluoride removal by 1 and 2 is due to an entropy effect rather than enthalpy change. The present study provides a new insight into the design of MOFs for adsorption applications and water remediation.Acknowledgements
This study was partially supported by the National Natural Science Foundation of China (NSFC 21501003), the Natural Science Foundation of Anhui Province (1608085QB27), the China Postdoctoral Science Foundation funded project (2015M581973) and the City Social Science and Technology Development Program of Dongguan (Grant 2016108101005) and Science Foundation funded project of Guangdong Medical University (Z2016001 and M2016023). We thank Professor S. Ng and Professor Stuart R. Batten for discussion and help.Notes and references
- S. Kanrar, S. Debnath, P. De, K. Parashar, K. Pillay, P. Sasikumar and U. C. Ghosh, Chem. Eng. J., 2016, 306, 269 CrossRef CAS
.
- J. G. Cai, Y. Y. Zhang, B. C. Pan, W. M. Zhang, L. Lv and Q. X. Zhang, Water Res., 2016, 102, 109 CrossRef CAS PubMed
- K. S. Prasad, Y. Amin and K. Selvaraj, J. Hazard. Mater., 2014, 276, 232 CrossRef CAS PubMed
- A. Dhillon and D. Kumar, J. Mater. Chem. A, 2015, 3, 4215 CAS
- L. Chen, K. S. Zhang, J. Y. He, W. H. Xu, X. J. Huang and J. H. Liu, Chem. Eng. J., 2016, 285, 616 CrossRef CAS
- S. V. Jadhav, E. Bringas, G. D. Yadav, V. K. Rathod, I. Ortiz and K. V. Marathe, J. Environ. Manage., 2015, 162, 306 CrossRef CAS PubMed
- S. P. Suriyaraj and R. Selvakumar, RSC Adv., 2016, 6, 10565 RSC
- A. Dhillon, M. Nair, S. K. Bhargava and D. Kumar, J. Colloid Interface Sci., 2015, 457, 289 CrossRef CAS PubMed
- W.-Y. Li, J. Liu, H. Chen, Y. Deng, B. Zhang, Z. Wang, X. Zhang and S. Hong, Chem. Eng. J., 2013, 225, 865 CrossRef CAS
- L. H. Velazquez-Jimenez, R. H. Hurt, J. Matos and J. R. Rangel-Mendez, Environ. Sci. Technol., 2014, 48, 1166 CrossRef CAS PubMed
- L. Y. Deng, Y. W. Liu, T. L. Huang and T. Sun, Chem. Eng. J., 2016, 287, 83 CrossRef CAS
- L. Chai, Y. Wang, N. Zhao, W. Yang and X. You, Water Res., 2013, 47, 4040 CrossRef CAS PubMed
- X. M. Wu, Y. Zhang, X. M. Dou, B. Zhao and M. Yang, Chem. Eng. J., 2013, 223, 364 CrossRef CAS
- Y. Yu and J. P. Chen, J. Mater. Chem. A, 2014, 2, 8086 CAS
- S. X. Dong and Y. L. Wang, Water Res., 2016, 88, 852 CrossRef CAS PubMed
- C. H. Wang, X. L. Liu, N. K. Demir, J. P. Chen and K. Li, Chem. Soc. Rev., 2016, 45, 5107 RSC
- Y. J. Cui, B. Li, H. J. He, W. Zhou, B. L. Chen and G. D. Qian, Acc. Chem. Res., 2016, 49, 483 CrossRef CAS PubMed
- P. Hu, J. V. Morabito and C. K. Tsung, ACS Catal., 2014, 4, 4409 CrossRef CAS
-
(a) M. I. Nandasiri, S. R. Jambovane, B. P. McGrail, H. T. Schaef and S. K. Nune, Coord. Chem. Rev., 2016, 311, 38 CrossRef CAS
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Feréy, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232 CrossRef CAS PubMed
- B. Wang, X. L. Lv, D. W. Feng, L. H. Xie, J. Zhang, M. Li, Y. B. Xie, J. R. Li and H. C. Zhou, J. Am. Chem. Soc., 2016, 138, 6204 CrossRef CAS PubMed
- A. J. Howarth, M. J. Katz, T. C. Wang, A. E. Platero-Prats, K. W. Chapman, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 7488 CrossRef CAS PubMed
- N. A. Khan, Z. Hasan and S. H. Jhung, J. Hazard. Mater., 2013, 244, 444 CrossRef PubMed
- A. Chakraborty, S. Bhattacharyya, A. Hazra, A. C. Ghosh and T. K. Maji, Chem. Commun., 2016, 52, 2831 RSC
- L. Li, S. Xiang, S. Cao, J. Zhang, G. Ouyang, L. Chen and C. Y. Su, Nat. Commun., 2013, 4, 1774 CrossRef PubMed
- K. Y. A. Lin, H. T. Yang, C. Petit and F. K. Hsu, Chem. Eng. J., 2014, 249, 293 CrossRef CAS
- N. A. Khan and S. H. Jhung, Angew. Chem., Int. Ed., 2012, 51, 1198 CrossRef CAS PubMed
- S. Karmakar, J. Dechnik, C. Janiak and S. De, J. Hazard. Mater., 2016, 303, 10 CrossRef CAS PubMed
- K. Y. A. Lin, Y. T. Liu and S. Y. Chen, J. Colloid Interface Sci., 2016, 461, 79 CrossRef CAS PubMed
- F. Ke, G. Luo, P. R. Chen, J. Jiang, Q. Y. Yuan, H. M. Cai, C. Y. Peng and X. C. Wan, J. Porous Mater., 2016, 23, 1065 CrossRef CAS
- N. Zhang, X. Yang, X. Yu, Y. Jia, J. Wang, L. Kong, Z. Jin, B. Sun, T. Luo and J. Liu, Chem. Eng. J., 2014, 252, 220 CrossRef CAS
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 7, 3 CrossRef PubMed
- J. Q. Liu, G. P. Li, W. C. Liu, Q. L. Li, B. H. Li, R. W. Gable, L. Hou and S. R. Batten, ChemPlusChem, 2016, 81, 1299 CrossRef CAS
- Y. Liu, Q. Fan, S. X. Wang, Y. L. Liu, A. L. Zhou and L. Fan, Chem. Eng. J., 2016, 306, 174 CrossRef CAS
- E. Haque, V. Lo, A. I. Minett, A. T. Harris and T. L. Church, J. Mater. Chem. A, 2014, 2, 193 CAS
Footnotes |
| † Electronic supplementary information (ESI) available: TGA, IR, XPS, PXRD, experiments of adsorption kinetics and thermodynamics. CCDC 1518722. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ce00291b |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |