
Adjacent N→O and C–NH2 groups — a highly efficient amphoteric structure for energetic materials resulting from tautomerization proved by crystal engineering†
Zhicun
Feng
 a,
Yu
Zhang
a,
Yanan
Li
b and
Kangzhen
Xu
*a
a,
Yu
Zhang
a,
Yanan
Li
b and
Kangzhen
Xu
*a
aSchool of Chemical Engineering & Integrated Military-Civilian Innovation Center for Energetic Materials, Northwest University, Xi'an 710069, China. E-mail: xukz@nwu.edu.cn
bXi'an Modern Chemistry Research Institute, Xi'an 710065, China
First published on 19th January 2021
Abstract
Existing amphoteric energetic materials (EMs) are very scarce, and their amphoteric structures are difficult to reproduce and protonate. Herein, adjacent N→O and C–NH2 groups (O←N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–NH2) were found to be a highly efficient and fairly balanced amphoteric energetic structure for EMs by crystal engineering and are the first discovered amphoteric structure caused by tautomerization to the best of our knowledge. As the O←NC–NH2 structure has existed in many synthesized nitrogen-rich EMs, our work will greatly enrich amphoteric EMs.
C–NH2) were found to be a highly efficient and fairly balanced amphoteric energetic structure for EMs by crystal engineering and are the first discovered amphoteric structure caused by tautomerization to the best of our knowledge. As the O←NC–NH2 structure has existed in many synthesized nitrogen-rich EMs, our work will greatly enrich amphoteric EMs.
Energetic materials (EMs), such as explosives, propellants and pyrotechnics, play an important role in the fields of spaceflight, national defense and firework shows.1,2 To meet the increasingly diversified demands for EMs, the efficiency of developing new EMs needs to be improved. Because pairing different energetic ions by simple ion exchange reactions to synthesize energetic salts is a very efficient way to discover new EMs in large quantities, many nonionic EMs, especially nitrogen-rich EMs,3–9 are often further employed as ion precursors.6,8,10–18 The key to ionizing EMs is their acid–base properties. Acidic and alkaline EMs can be deprotonated and protonated to give the corresponding energetic anions and cations, respectively. Typical examples of deprotonation and protonation of EMs are summarized in Scheme S1.† Most EMs only can be deprotonated or protonated because they only show acidity or alkalinity. Amphoteric EMs which can produce both energetic anions and energetic cations are very scarce at present. To the best of our knowledge, only five amphoteric energetic compounds were reported in the literature (Scheme S2†).19–22,43,44 Moreover, although their amphoteric mechanisms were revealed by their crystal structures clearly, their amphoteric structures are difficult to reproduce, and their protonation requires very strong acids (like CF3SO3H and HClO4) or severe conditions (like the use of 90% HNO3 and HCl gas). Thus, to facilitate the synthesis of energetic salts, new highly efficient and easily available amphoteric structures for EMs need to be found.
Here, we report that we found a highly efficient amphoteric structure for EMs by crystal engineering while exploring the synthesis of 2,4,6-triamino-1,3,5-triazine-1,3,5-trioxide (MTO). MTO was suggested as a candidate for green EMs by Klapötke in 2014 but has not yet been synthesized.23,24 As shown in Scheme 1, we tried oxidizing melamine to synthesize MTO. Although the attempts to synthesize MTO failed, we synthesized two analogues of MTO, namely, 2,4,6-triamino-1,3,5-triazine-1,3-dioxide (1) and 4,6-diamino-3-hydroxy-2-oxo-2,3-dihydro-1,3,5-triazine-1-oxide (2), unexpectedly. Among them, 1 was first reported by Song et al. recently,25 and was found to be able to self-assemble with oxidant molecules (like H2O2 and fuming nitric acid) to obtain self-assembled energetic materials with better properties. 1 and 2 were synthesized in the same reaction system at different temperatures. 2 is actually a product from the hydrolysis of the amino group between two N→O coordinate bonds of 1 at higher temperature. To determine the tautomeric tendencies between 1 and 1′, and 2 and 2′ shown in Scheme 1, the crystal structures of 1·4H2O and 2·0.5H2O were obtained (Fig. 1). 1 is only soluble in water at high temperature, so the single crystals of 1·4H2O were obtained by slowly cooling its hot water solution (Fig. S1a†). However, 2 is insoluble in water and common organic solvents. The single crystal growth of 2 was unexpected and will be depicted together with that of metal salts of 2 later. From the difference Fourier maps of the X-ray diffraction data of single crystals of 1·4H2O and 2·0.5H2O, we found that there were two strong H atom signals around every amino N atom but no H atom signal around O atoms for 1, and there was a strong H atom signal around O(2) but no H atom signal around O(3) for 2 (Fig. 1). Moreover, the C(1)–O(3) bond distance of 2 (1.220 Å) agrees well with the normal CO bond distance (1.19–1.23 Å).26 Therefore, the crystal structures indicated that the adjacent N→O and C–NH2 groups (the O←NC–NH2 structure) of 1 can persist stably, whereas the adjacent N→O and C–OH groups (the O←NC–OH structure) of 2′ tautomerized into the HO–N–CO structure of 2. The thermodynamic stability of the tautomers of 1 and 2 was further supported by quantum chemical calculations (see ESI† Note 1). The molecular total energies (sum of the electronic and thermal energies) at 298.15 K of 1 and 2 were calculated to be 40.77 and 29.69 kJ mol−1 less than those of 1′ and 2′, respectively, which also supported that the O←NC–NH2 and HO–N–CO structures are more stable than the corresponding HO–N–CNH and O←NC–OH tautomeric structures, respectively.
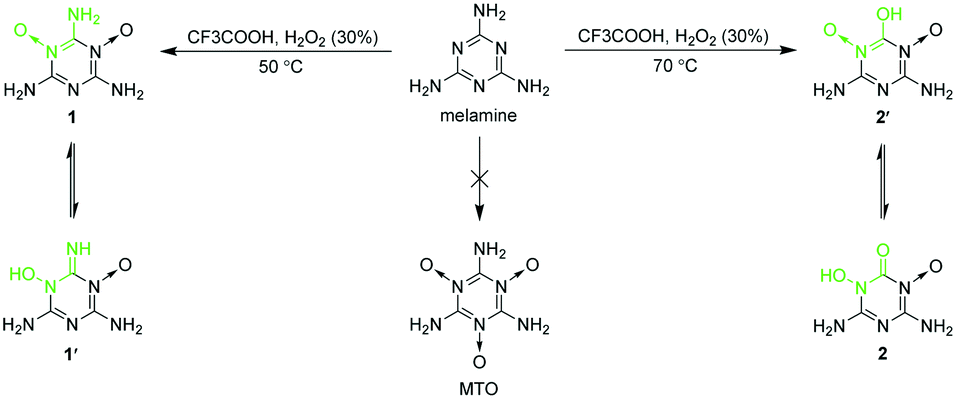 | ||
| Scheme 1 N-Oxides of melamine. | ||
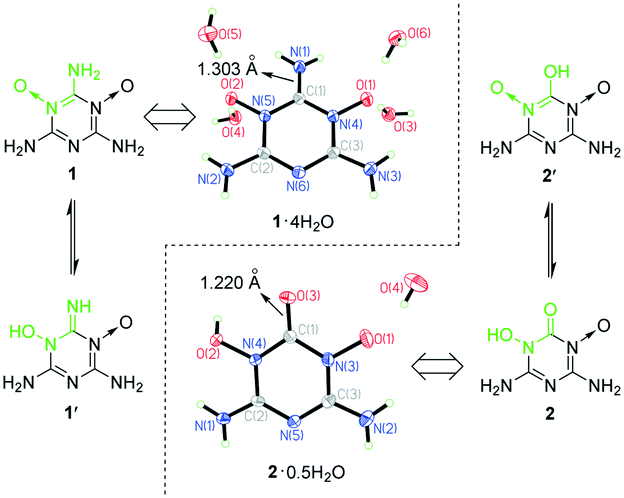 | ||
| Fig. 1 Crystal structures of 1·4H2O and 2·0.5H2O showing their tautomeric tendencies. | ||
To explain why 1 was difficult to further oxidize to form MTO and why the amino group between its two N→O bonds was easy to hydrolyze at high temperature, its crystal structure was used to study the electron transfer of melamine coordinated with two O atoms (see ESI† Note 2).27–29 As shown in Fig. 2, it is obvious that the electron density around N(6) decreased widely after N(4) and N(5) coordinated with O(1) and O(2), respectively, which resulted in a considerable decrease of its coordination ability. Thus, the difficulty level of oxidizing N(6) of 1 to form another N→O coordinate bond increased, which made MTO difficult to obtain. In addition, the overall electron density around C(1) dropped more than those around C(2) and (C3), which made it more vulnerable to attacks from nucleophiles, so the amino group N(1)H2 of 1 was easier to hydrolyze than its amino groups N(2)H2 and N(3)H2, resulting in the formation of 2 at high temperature.
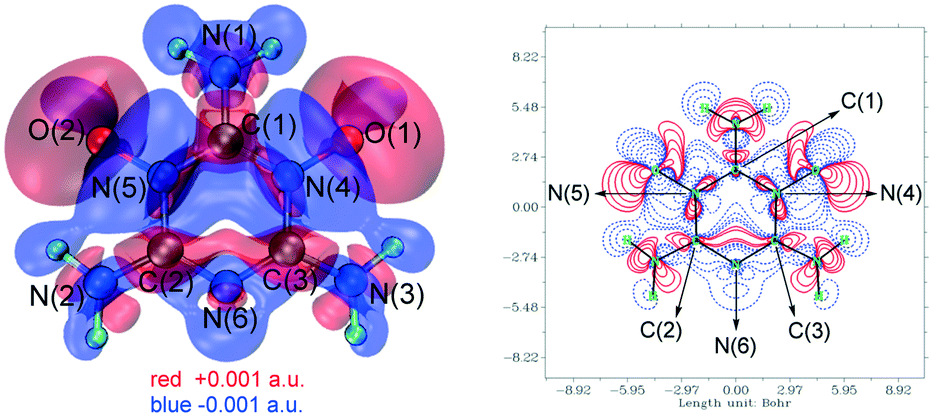 | ||
| Fig. 2 Isosurface (left) and contour (right) maps of the electron density difference between melamine and 1 (the red and blue parts represent the regions in which the electron density increases and decreases after melamine coordinated to two O atoms). | ||
Although 1 and 2 were hardly soluble in water at room temperature, we found that they could dissolve in both acidic and alkaline solutions at room temperature, which betokened that they are probably amphoteric EMs. To verify our guess, we tried using nitric acid, NaOH and KOH to protonate and deprotonate 1 and 2. As a result, we facilely obtained sodium salts (1a and 2a), potassium salts (1b and 2b) and nitrate salts (1c, 1d and 2c) of 1 and 2 (Scheme 2). To accurately determine the amphoteric mechanisms of 1 and 2 as shown in Scheme 2, the crystal structures of 1a·5H2O, 1b, 2a·4H2O, 2b·2.5H2O, 1c·H2O, 1d·H2O and 2c were determined (Fig. 3 and 4). The single crystals of 1c·H2O, 1d·H2O and 2c were obtained from the cooling process when 1c, 1d and 2c were synthesized (Fig. S1a†). Although 1a, 2a, 1b and 2b only can dissolve in water, their single crystals were not directly grown by volatilization of their water solutions, because 1′− and 2− can combine with H+ ionized from H2O to re-form insoluble 1 and 2, which resulted in the formation of many crystals of 1·2H2O and the abovementioned 1·4H2O and 2·0.5H2O in the water solutions of 1a, 2a, 1b and 2b (Fig. S1b†). The hydrolysis properties of 1′− can also be proved using the NMR spectra (see ESI† Note 3). The single crystals of 1a·5H2O, 1b, 2a·4H2O and 2b·2.5H2O were grown by a vapor diffusion method using the corresponding NaOH or KOH aqueous solutions of 1a, 2a, 1b and 2b (Fig. S1c†).
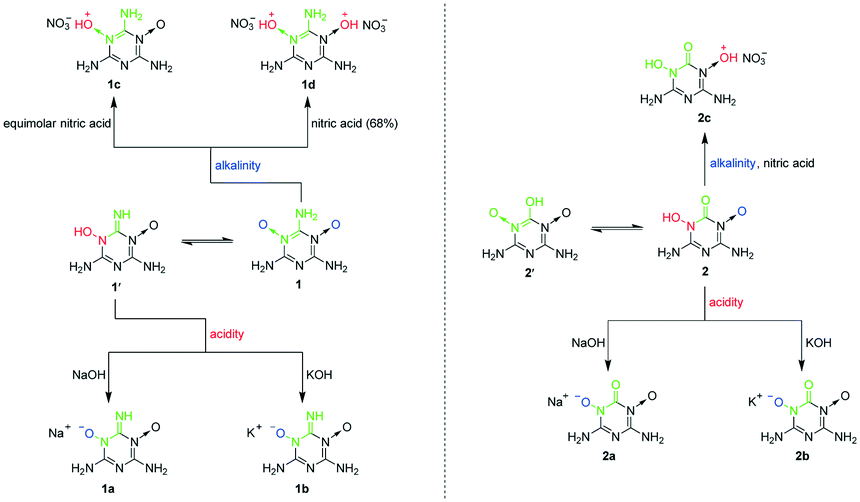 | ||
| Scheme 2 Protonation and deprotonation of 1 and 2. | ||
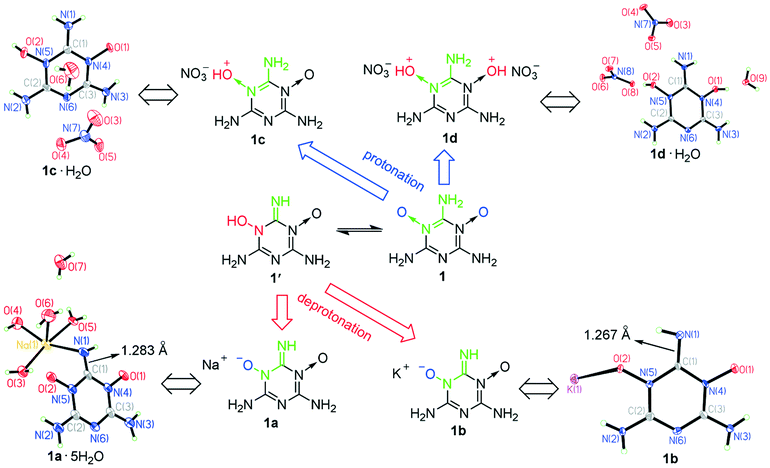 | ||
| Fig. 3 Crystal structures of the energetic salts of 1 showing its amphoteric mechanism. | ||
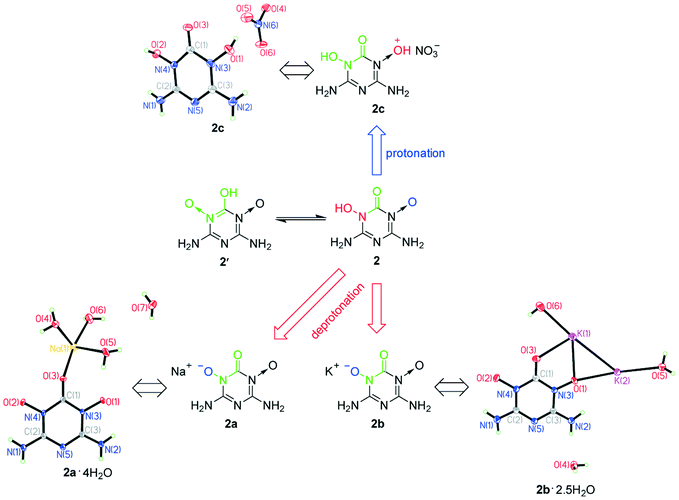 | ||
| Fig. 4 Crystal structures of the energetic salts of 2 showing its amphoteric mechanism. | ||
After analysing the signals of H atoms from the collected X-ray diffraction data of 1c·H2O and 1d·H2O (Fig. 3), we found that all the protons from nitric acid were accepted by the O atoms of the N→O bonds, so the N→O bonds are alkaline. Moreover, because the two N→O bonds of 1 make it a diacidic base, not only dinitrate salt 1d reported by Song et al.25 but also a new mononitrate salt of 1 (1c) was synthesized using equimolar diluted nitric acid. Thus, the O←NC–NH2 structure can be easily protonated. As the traditional method of obtaining nitrogen-rich energetic cations was the protonation of N atoms (Scheme S1†), we found that this way of protonating O atoms of N→O coordinate bonds is a new efficient method of obtaining nitrogen-rich energetic cations. According to the X-ray diffraction data of 1a·5H2O and 1b (Fig. 3), there was only one H atom signal around their N(1) atoms, and the C(1)–N(1) bond distance of 1.303 Å in 1·4H2O shortened to 1.283 and 1.267 Å in 1a·5H2O and 1b, respectively, which indicated that the O←NC–NH2 structure tautomerized into the HO–N–CNH structure during the deprotonation. Therefore, although the O←NC–NH2 structure is more stable than its tautomeric structure HO–N–CNH and cannot be deprotonated directly, the deprotonation of its tautomeric structure under alkaline conditions can lead to the tautomeric equilibrium between O←NC–NH2 and HO–N–CNH continuously moving in the HO–N–CNH direction. As a result, the O←NC–NH2 structure can produce a large number of energetic anions in the form of (O–N–CNH)− under alkaline conditions. According to the formation of 1a–1d, the O←NC–NH2 structure can be easily protonated and deprotonated using common acids and alkalis (diluted nitric acid, NaOH and KOH) to give the corresponding nitrogen-rich energetic cations and anions, respectively, so it is a highly efficient amphoteric structure for constructing amphoteric EMs. As far as we know, it is the first discovered amphoteric structure caused by tautomerization. In addition, we found that the O←NC–NH2 structure has existed in many synthesized nitrogen-rich EMs (Scheme S3†),25,30–35 so the O←NC–NH2 structure is easy to construct and has high potential to greatly enrich amphoteric EMs.
To further quantitatively characterize the acidity and alkalinity of the tautomeric structures O←NC–NH2 and HO–N–CNH, the first basic dissociation constant (Kb1) of 1 and the acidic dissociation constant (Ka) of 1′ were evaluated by determining the pH values of the water solutions of their salts (see ESI† Note 4). Kb1 of 1 and Ka of 1′ were measured to be 3.20 × 10−11 and 1.19 × 10−10, respectively. The alkalinity of 1 is slightly weaker than that of aniline (Kb = 3.98 × 10−10), and the acidity of 1′ is comparable to that of phenol (Ka = 1.02 × 10−10).36 The close basic and acidic dissociation constants of 1 and 1′ show that the alkalinity of the O←NC–NH2 structure is close to the acidity of its tautomeric structure HO–N–CNH. Thus, the O←NC–NH2 structure is a fairly balanced amphoteric structure. In addition, the weak acidity of 1′ makes its conjugate base 1′− a strong base, and the O←NC–NH2 structure is more thermodynamically stable than its tautomeric structure HO–N–CNH, so it is difficult to further deprotonate 1′−, although there is still a O←NC–NH2 structure in its structure.
As shown in Fig. 4, the N→O bond of 2 was also protonated using nitric acid. However, only the mononitrate salt 2c was obtained because there was only one N→O bond in 2 after the other tautomerized into the HO–N–CO structure with the C–OH group. The HO–N–CO structure of 2 was also deprotonated under alkaline conditions because there was no H atom signal around the O(1) and O(2) atoms of 2a·4H2O and 2b·2.5H2O according to their X-ray diffraction data (Fig. 4), resulting in the formation of energetic anions in the form of (O–N–CO)−. Because the O←NC–OH structure mainly exists as the more stable HO–N–CO structure, it loses the ability to be protonated. Thus, amphoteric energetic compounds cannot be achieved using the O←NC–OH structure alone.
Groups with negative and positive surface electrostatic potentials (ESPs) have potential to accept and give protons, respectively.37 To promote understanding of the acid–base properties of the O←NC–NH2 and HO–N–CO structures, the ESPs of 1, 1′, 2 and 2′ were calculated (see ESI† Note 5).27–29 As shown in Fig. 5, the ESPs around N(1)H2 and O(2) of 1 are positive and negative, respectively, indicating that N(1)H2 and O(2) are inclined to give and accept protons, respectively. Such inclinations should provide some kind of driving force for the tautomerization between O←NC–NH2 and HO–N–CNH. After the O(2) atom of 1 participated in the tautomerization, the ESPs around it changed in the positive direction, which gave 1′ the potential to be deprotonated. For 2′, the ESPs around O(2) are lower than those of 1, so the isomerization from 2′ to 2 proceeds more easily than that from 1 to 1′. The ESPs around O(1) and O(2) of 2 are at the negative and positive extremes, respectively, which gives 2 amphoteric properties.
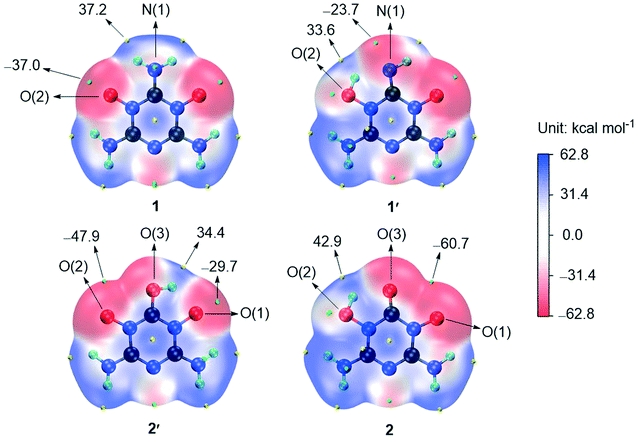 | ||
| Fig. 5 ESPs (at 0.001 a.u.) of 1, 1′, 2 and 2′ showing the maximum (yellow pellets) and minimum (green pellets) points. | ||
The structure refinement results of all the crystals are given in Tables S1 and S2.† Affected by the strong electrostatic attraction of the O atoms of N→O bonds and nitrate ions, 1·4H2O, 1c·H2O, 1d·H2O and 2·0.5H2O can easily attract lattice water to bond with the O atoms using hydrogen bonds (Fig. S2†). For 1a·5H2O, 2a·4H2O and 2b·2.5H2O, because the ionic radii of their metal ions, especially Na+, are short, the resulting strong electric fields in water caused much coordinated water in their crystal structures. All the bond distances between the C and N atoms of the synthesized nine compounds are in the range of 1.267–1.384 Å, which correspond to a conjugated double CN bond (1.34–1.38 Å),26 indicating that their triazine moieties exhibit extensive π conjugations.
Both 1 and 2 are planar molecules, and the molecular packing of their crystals consists of wave-shaped layers with extensive intermolecular hydrogen bonds and π–π molecular stacking (Fig. S3†). Sodium salts 1a·5H2O and 2a·4H2O and potassium salts 1b and 2b·2.5H2O are complexes. Among them, 2a·4H2O and 2b·2.5H2O are one-dimensional (1D) metal–organic frameworks (MOFs) (Fig. S4†), and 1b is a three-dimensional (3D) MOF (Fig. 6a). In 1b, every K+ is five-coordinated by four 1′−, and every 1′− coordinates with four K+, which makes K+ and 1′− connected as a 3D supramolecular framework with π–π and hydrogen bond interactions (Fig. S5†). Nitrate salts 1d·H2O and 2c exhibit similar 3D graphite-like layered packing, whereas nitrate salt 1c·H2O, like 1·4H2O and 2·0.5H2O, exhibits wave-layered packing (Fig. 6b). There are seven kinds of hydrogen bonds within the two-dimensional (2D) molecular sheets of 1c·H2O and 1d·H2O. Then their 2D layer structures are connected by three kinds of hydrogen bonds between the layers. For crystal 2c, there are three kinds of hydrogen bonds within the 2D molecular sheets and two kinds of hydrogen bonds between the sheets. The detailed hydrogen bond information of all the crystals is summarized in Table S3.†38,39
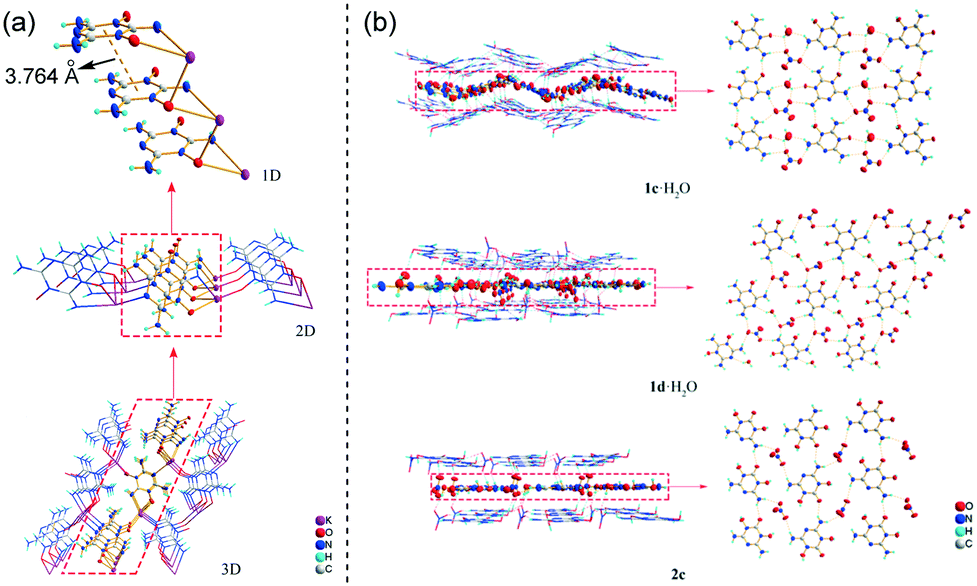 | ||
| Fig. 6 (a) Structure of 3D MOF 1b showing π–π stacking (the dashed lines); (b) 3D layered packing and 2D molecular sheets of crystals 1c·H2O, 1d·H2O and 2c showing hydrogen bonds (the dashed lines). | ||
The constant volume combustion energies of all the newly synthesized energetic compounds were measured by oxygen bomb calorimetry, and then were used to calculate their heat of formation (ΔfHm) (see ESI† Note 6). Their physical and energetic properties, including decomposition temperature (Td), detonation pressure (P), detonation velocity (vD) and impact sensitivity (IS), are summarized in Table S4† in detail. 1a (Td = 325 °C, P = 20.7 GPa, vD = 6572 m s−1, IS > 23.5 J) and 2 (Td = 293 °C, P = 22.8 GPa, vD = 6976 m s−1, IS > 23.5 J) show very high thermal stability and low impact sensitivity, and possess detonation properties comparable to 1 (P = 20.7 GPa, vD = 6924 m s−1)25 and 2,4,6-trinitrotoluene (TNT) (P = 19.4 GPa, vD = 6915 m s−1).40 Metal salts 1b, 2a and 2b (Td = 287–317 °C, P = 14.9–16.3 GPa, vD = 5606–5767 m s−1, IS > 23.5 J, ΔfHm ≤ 215.6 kJ mol−1) have thermal stability and impact sensitivity similar to sodium salt 1a, but their detonation properties are lower than those of 1a (ΔfHm = −164.2 kJ mol−1) because of their lower negative ΔfHm. Such differences between the ΔfHm values may mainly come from the differences of types and quantities of the coordinate bonds between the metal salts. The strong π conjugations on the triazine moieties and π–π molecular stacking in the crystal structures, which can stabilize molecules,41 should be related with the high stability of the above heat-resistant insensitive compounds. The stability and detonation properties of mononitrate salts 1c (Td = 226 °C, P = 24.1 GPa, vD = 7459 m s−1, IS = 6.9 J) and 2c (Td = 194 °C, P = 25.3 GPa, vD = 7503 m s−1, IS = 6.9 J) are lower and better than those of the metal salts, respectively, because of the introduction of the energetic nitrate ions. 1c and 2c exhibit higher thermal stability, more appropriate impact sensitivities and slightly better detonation performance than 2-diazo-4,6-dinitrophenol (DDNP) (P = 24.2 GPa, vD = 6900 m s−1, Td = 157 °C, IS = 1 J),42 and do not contain chlorine or heavy metals, so they have good potential as green primary explosives. Compared with 1c and 2c, although dinitrate salt 1d (Td = 180 °C, P = 34.2 GPa, vD = 8900 m s−1, IS = 14 J)25 exhibits poorer thermal stability, it has better detonation properties and lower impact sensitivity because of the higher energetic nitrate ion content and the resulting more electrostatic attraction. In addition, the above layered packing aided by extensive intermolecular hydrogen bond interactions, which can increase the packing compactness and strengthen anisotropy of EMs to facilitate their ready shear slide and low mechanical sensitivity,41 is closely related to the low impact sensitivity of the nitrate salts. The facile synthesis of these series of energetic salts with different properties using common inorganic acids and alkalis shows that our ways of obtaining amphoteric EMs are easy and efficient.
Conclusions
In summary, the adjacent N→O and C–NH2 groups (the O←NC–NH2 structure) were found to be a highly efficient and fairly balanced amphoteric structure for EMs. The amphoteric mechanism of the O←NC–NH2 structure was revealed by crystal engineering and quantum chemical calculations in detail. Its alkalinity is from the N→O coordinate bond, and its acidity is from the N–OH group resulting from its tautomeric structure HO–N–CNH. To the best of our knowledge, this is the first discovered amphoteric structure caused by tautomerization. As the O←NC–NH2 structure has existed in many synthesized nitrogen-rich EMs, and previously reported amphoteric EMs were very scarce and were difficult to protonate, our work will greatly enrich amphoteric EMs. In addition, the crystal analysis showed that amphoteric EMs cannot be achieved using the similar O←NC–OH structure alone because its tautomeric structure HO–N–CO is more stable than itself. Two amphoteric nitrogen-rich EMs were found in this paper, and their seven energetic salts with different properties were synthesized. Their anions and cations can be combined with more energetic cations and anions (such as hydrazinium cations, hydroxylammonium cations, dinitramide anions et al.), respectively, to obtain more promising nitrogen-rich energetic salts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21673178) and the National Defense Scientific Research Project.Notes and references
-
H. H. Krause, Energetic Materials, VCH, Weinheim, 2005 Search PubMed
.
-
T. M. Klapötke, Chemistry of High-Energy Material, Walter de Gruyter GmbH & Co. KG, Berlin/New York, 2011 Search PubMed
- D. E. Chavez, M. A. Hiskey and R. D. Gilardi, Angew. Chem., Int. Ed., 2000, 39, 1791–1793 CrossRef CAS
- D. E. Chavez, M. A. Hiskey and D. L. Naud, Propellants, Explos., Pyrotech., 2004, 29, 209–215 CrossRef CAS
- G.-H. Tao, B. Twamley and J. M. Shreeve, J. Mater. Chem., 2009, 19, 5850–5854 RSC
- T. M. Klapötke and T. G. Witkowski, Propellants, Explos., Pyrotech., 2015, 40, 366–373 CrossRef
- T. Wang, J. Zhou, Q. Zhang, L. Zhang, S. Zhu and Y. Li, New J. Chem., 2020, 44, 1278–1284 RSC
- J.-T. Wu, J. Xu, W. Li and H.-B. Li, Propellants, Explos., Pyrotech., 2020, 45, 536–545 CrossRef CAS
- W. Huang, Y. Tang, G. H. Imler, D. A. Parrish and J. M. Shreeve, J. Am. Chem. Soc., 2020, 142, 3652–3657 CrossRef CAS
- J. C. Oxley, J. L. Smith and H. Chen, Thermochim. Acta, 2002, 384, 91–99 CrossRef CAS
- R. P. Singh, R. D. Verma, D. T. Meshri and J. M. Shreeve, Angew. Chem., Int. Ed., 2006, 45, 3584–3601 CrossRef CAS
- T. M. Klapötke, C. M. Sabaté and J. Stierstorfer, Z. Anorg. Allg. Chem., 2008, 634, 1867–1874 CrossRef
- T. M. Klapötke, C. M. Sabaté and J. M. Welch, Z. Anorg. Allg. Chem., 2008, 634, 857–866 CrossRef
- M. Göbel, K. Karaghiosoff, T. M. Klapötke, D. G. Piercey and J. Stierstorfer, J. Am. Chem. Soc., 2010, 132, 17216–17226 CrossRef
- H. Huang, Z. Zhou, L. Liang, J. Song, K. Wang, D. Cao, C. Bian, W. Sun and M. Xue, Z. Anorg. Allg. Chem., 2012, 638, 392–400 CrossRef CAS
- T. M. Klapötke, P. C. Schmid, S. Schnell and J. Stierstorfer, Chem. – Eur. J., 2015, 21, 9219–9228 CrossRef
- C. Bian, X. Dong, X. Zhang, Z. Zhou, M. Zhang and C. Li, J. Mater. Chem. A, 2015, 3, 3594–3601 RSC
- P. He, J.-G. Zhang, X. Yin, J.-T. Wu, L. Wu, Z.-N. Zhou and T.-L. Zhang, Chem. – Eur. J., 2016, 22, 7670–7685 CrossRef CAS
- T. M. Klapötke, M. Stein and J. Stierstorfer, Z. Anorg. Allg. Chem., 2008, 634, 1711–1723 CrossRef
- T. M. Klapötke and J. Stierstorfer, Dalton Trans., 2009, 643–653 RSC
- T. T. Vo and J. M. Shreeve, J. Mater. Chem. A, 2015, 3, 8756–8763 RSC
- Y. Xu, P. Wang, Q. Lin, Y. Du and M. Lu, Sci. China Mater., 2019, 62, 751–758 CrossRef
- T. M. Klapötke, personal communication, 2014.
- T. Zhou, S. V. Zybin, W. A. Goddard, T. Cheng, S. Naserifar, A. Jaramillo-Botero and F. Huang, Phys. Chem. Chem. Phys., 2018, 20, 3953–3969 RSC
- S. Song, Y. Wang, W. He, K. Wang, M. Yan, Q. Yan and Q. Zhang, Chem. Eng. J., 2020, 395, 125114 CrossRef CAS
-
X. M. Chen and J. W. Cai, Single-Crystal Structure Analysis Principles and Practices, Science Press, Beijing, 2007 Search PubMed
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS
- T. Lu and F. Chen, J. Mol. Graphics Modell., 2012, 38, 314–323 CrossRef CAS
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS
- H.-H. Licht and H. Ritter, J. Energ. Mater., 1994, 12, 223–235 CrossRef CAS
- H. Ritter and H. H. Licht, J. Heterocycl. Chem., 1995, 32, 585–590 CrossRef CAS
- D. Fischer, T. M. Klapötke and J. Stierstorfer, Eur. J. Inorg. Chem., 2014, 2014, 5808–5811 CrossRef CAS
- H. Wei, H. Gao and J. M. Shreeve, Chem. – Eur. J., 2014, 20, 16943–16952 CrossRef CAS
- C. Ma, Z. Liu, X. Xu and Q. Yao, Chin. J. Org. Chem., 2014, 34, 1288–1299 CrossRef CAS
- Y. Wang, Y. Liu, S. Song, Z. Yang, X. Qi, K. Wang, Y. Liu, Q. Zhang and Y. Tian, Nat. Commun., 2018, 9, 2444 CrossRef
-
J. A. Dean, Lange's Handbook of Chemistry, McGraw-Hill, Inc., New York, 15th edn, 1999 Search PubMed
- J. S. Murray and P. Politzer, WIREs Comput. Mol. Sci., 2011, 1, 153–163 CrossRef CAS
- J. M. A. Robinson, D. Philp, K. D. M. Harris and B. M. Kariuki, New J. Chem., 2000, 24, 799–806 RSC
- A. K. Ghosh, A. Hazra, A. Mondal and P. Banerjee, Inorg. Chim. Acta, 2019, 488, 86–119 CrossRef CAS
- S.-L. Chen, Z.-R. Yang, B.-J. Wang, Y. Shang, L.-Y. Sun, C.-T. He, H.-L. Zhou, W.-X. Zhang and X.-M. Chen, Sci. China Mater., 2018, 61, 1123–1128 CrossRef CAS
- C. Zhang, F. Jiao and H. Li, Cryst. Growth Des., 2018, 18, 5713–5726 CrossRef CAS
-
R. Matyáš and J. Pachman, Primary Explosives, Springer, Heidelberg, 2013 Search PubMed
- M. von Denffer, T. M. Klapötke, G. Kramer, G. Spieß, J. M. Welch and G. Heeb, Propellants, Explos., Pyrotech., 2005, 30, 191–195 CrossRef CAS
- G.-H. Tao, Y. Guo, Y.-H. Joo, B. Twamley and J. M. Shreeve, J. Mater. Chem., 2008, 18, 5524–5530 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1967830, 1967836, 1967846, 1968244, 1968247, 1968249, 1972805, 1972807, 1972810 and 1977527. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce01427c |
| This journal is © The Royal Society of Chemistry 2021 |