
A user-friendly application for predicting the outcome of co-crystallizations†
Nandini
Sarkar
 a,
Joydeep
Mitra
b,
Molly
Vittengl
c,
Lexi
Berndt
a and
Christer B.
Aakeröy
*a
a,
Joydeep
Mitra
b,
Molly
Vittengl
c,
Lexi
Berndt
a and
Christer B.
Aakeröy
*a
aDepartment of Chemistry, Kansas State University, 213 CBC Building, 1212 Mid Campus Dr North, Manhattan, KS 66506-0401, USA. E-mail: aakeroy@ksu.edu; Fax: +785 532 6666; Tel: +785 532 6096
bDepartment of Computer Science, Kansas State University, Manhattan, Kansas 66506, USA
cDepartment of Chemistry, Truman State University, Kirksville, Missouri 63501, USA
First published on 30th September 2020
Abstract
An automated application, CoForm, was used for predicting the outcomes of attempted co-crystallizations between two active pharmaceutical ingredients, loratadine and desloratadine, and 41 potential co-formers from the general interest (OGI) list. The predictive abilities of the app were compared to structure-informatics tools based on hydrogen-bond propensity (HBP) and molecular complementarity (MC). The results indicate that CoForm delivered a success rate of 78% for both loratadine and desloratadine compared to 76% and 54%, respectively (HBP), and 39% and 22%, respectively (MC).
Introduction
Pharmaceutical companies invest vast resources for research and development of new and more effective drugs.1–3 The challenges of delivering a viable product are considerable and in addition to possessing optimal biological properties, a successful candidate must also present appropriate physicochemical/pharmacological properties such as solubility, stability, dissolution rate, bioavailability, and shelf life.4–6 A majority of compounds that are eliminated in this process fail due to sub-par physicochemical properties rather than to unacceptable toxicity.7 Solubility is one of the major issues in orally administrated drugs as inadequate aqueous solubility or dissolution rate lead to lower therapeutic effect. Many different approaches have been utilized to address this issue, such as nanocrystal formation, amorphization, salt formation, co-crystallization, and polymorph screens.8In the last two decades, co-crystallization technologies have emerged as an area of research involving high value organic crystalline solids. A pharmaceutical co-crystal is the result of a successful combination of an active pharmaceutical ingredient (API) and an appropriate molecular partner, the co-former. Unfortunately, finding molecules that can act as co-formers for a specific drug is generally based on combinatorial and extensive experimental co-crystal screens, which are time-consuming and expensive.9 One of the reasons why co-crystal synthesis has not yet transitioned into a widely utilized technology is partly due to challenges associated with finding molecules that are likely to form a new solid crystalline form with the API. Consequently, there is a need for cheaper, faster, and more reliable methods for predicting when a pair of molecules will form a co-crystal, and when they will not.
There are a handful of predictive methods for co-crystal formation in the literature. However, some of these methods are very complex and require in-depth knowledge of theoretical chemistry and quantum mechanical methods.10–13 Such methods also tend to be computationally expensive and less suitable for systematic screens. Other methods have employed combinations of data mining and structure-informatics, taking advantage of over a million crystal structures of small molecules in the Cambridge Structural Database (CSD).14–16 Thanks to the presence of reliable and properly curated data in the CSD, various structure-informatics methods such as hydrogen-bond propensity,9,17 hydrogen-bond coordination,18 and molecular complementarity19 developed by the Cambridge Crystallographic Data Centre (CCDC) have been applied to co-crystal prediction. One inherent problem with building a predictive tool on existing crystal structures is that only positive co-crystallization results are included, failed co-crystallizations can by definition not be included in any training data set. With this in mind, access to a new approach for accurately predicting the outcome of co-crystallization reactions based on both positive and negative experimental results could be of interest to a broad spectrum of the organic solid-state community.
The CoForm application
In order to address the wide-ranging needs for versatile protocols for co-crystal synthesis, we have developed an automated application for predicting the outcome of attempted co-crystallizations. The work was motivated by a need to streamline expensive and time-consuming experimental processes for finding a suitable co-former candidate for any given small-molecule API.CoForm is based on a mathematical model that compares the number of hydrogen-bond donors and acceptors of the target of interest with the number of hydrogen-bond donors and acceptors of a set of known compounds. Each target is associated with a list of co-formers with which it forms co-crystals (positive partners), and a list of co-formers with which it does not form co-crystals (negative partners). See ESI† Fig. S1 for a detailed description of the algorithm. The database for the known compounds is based on the outcome (as determined using infrared spectroscopy) of approximately 2000 attempted co-crystallizations.20–23 The quality of the predictions using CoForm is dependent on the compounds present in the database, however, the app can be customized to work with databases that are directly tailored to the type of target compounds and co-formers that a prospective user is specifically interested in. The automated algorithm is very fast and accessible through an easy-to-use desktop application. Moreover, users with relatively limited technical knowledge will be able to use the app and interpret the results. The current version of CoForm is based on a database that comprises 41 co-formers that are of general interest (OGI) for pharmaceutical co-crystals and an additional 50 co-formers, which are conventionally used in co-crystallization experiments (see ESI† Table S1).
CoForm is built using the Groovy programming language.24 Groovy was chosen because it is platform-independent and, therefore, the app can be used on all three major operating systems, i.e., Windows, Linux, and Mac OSX. Moreover, Groovy is a scripting language that allows quick prototyping of software.
CoForm requires two inputs from the user:
1. Number of hydrogen-bond donors (donor: molecule or molecular fragment X–H in which X is an electronegative atom such as N, O, and F).
2. Number of hydrogen-bond acceptors (acceptor: an electronegative element such as N, and O).
The name of the target for which co-crystals need to be predicted can also be incorporated to facilitate usability. The target name is simply providing a label/tag for the search but does not have any scientific meaning.
CoForm ranks the co-formers as ‘highly likely’, ‘likely’, and ‘least likely’ to produce a co-crystal with a specific target. The output is in the form of tables that can be exported as .csv files. The most likely and least likely lists, as the names suggest, correspond to the co-formers with the highest and lowest probability of forming a co-crystal, respectively. The likely lists consist of co-formers which were found to form co-crystals with compounds in the database in some cases and did not form in other cases. Since the co-crystallization outcomes are binary, we assigned the likely list of co-formers a ‘YES’ to co-crystallization. Although this will generate some false positives, this is more preferable than predictions of false negatives in the context of co-crystallization screens.
Validation study
In order to examine the accuracy of CoForm and its potential limitations in predicting co-crystallization outcomes, we carried out a systematic study where we matched the predicted results with the experimental co-crystallization outcomes for two known antihistamine drugs, loratadine, and desloratadine, Fig. 1. The targets were chosen as they have similar molecular backbone, but they present different hydrogen-bond donor acceptor ratios. The experimental part of the validation study involved attempted co-crystallizations, using solvent-assisted grinding, of both APIs against 41 co-formers on the OGI list (see ESI† Table S1).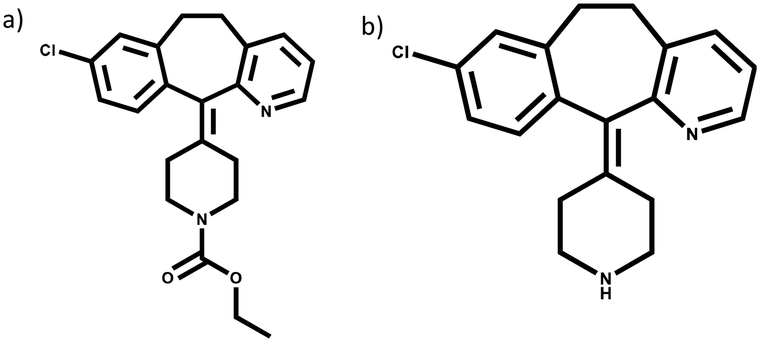 | ||
| Fig. 1 Molecular structures of a) loratadine; b) desloratadine. | ||
CoForm is a data-driven predictive application based on experimental data from attempted co-crystallization experiments which include both successful and unsuccessful reactions. In contrast, other structure-informatics analytical tools such as hydrogen-bond propensity (HBP)25 and molecular complementarity (MC)19 rely exclusively on existing crystallographic data, but both methods can be used for predicting co-crystallization outcomes (see ESI† Table S2). A comparison of the prediction outcomes of CoForm, HBP and MC methods was carried out on the same two molecules, loratadine and desloratadine. The accuracy of each method was determined by calculating the success rate, which is the number of predictions that match the experimental results over the total number of predictions (see ESI† S3–S6 for experimental and predicted co-crystallization screening outcomes).
The three methods gave the following success rate for predicting the outcome of 41 attempted co-crystallizations of loratadine: CoForm, 78%, HBP, 76%, and MC 39%. A summary of the results is displayed in a confusion matrix, Fig. 2.
 | ||
| Fig. 2 Correlation between the experimental and predicted outcomes for loratadine. | ||
30 of the 41 attempted co-crystallizations with loratadine produced a positive result and CoForm, HBP, and MC predicted these correctly at a success rate of 78%, 86%, and 26%, respectively. For the 11 reactions that did not produce a co-crystal, the three methods correctly predicted this with a success rate of 72% (CoForm), 45% (HBP), and 63% (MC).
A similar analysis of the predictions for co-crystallizations on desloratadine (again, 41 reactions were attempted) is given in Fig. 3.
 | ||
| Fig. 3 Correlation between the experimental and predicted outcomes for desloratadine. | ||
CoForm displays 89%, HBP 50%, and MC 16% prediction accuracy for successful co-crystallization outcomes, and for the failed attempts CoForm could not predict any of the five instances correctly, while HBP and MC both predicted 3/5 instances correctly.
Overall for both loratadine and desloratadine, CoForm produced higher success rates for the positive co-crystallization experiments. When comparing the ratio of successful to failed co-crystallization cases in our database, we found that there is a total of 1136 successful co-crystals and 649 failed co-crystals results. The positive outcomes account for 68% of the total number of attempted reactions which can help to explain why CoForm shows an imbalance for predicting positive versus negative outcomes. In Table 1, the overall success rates for the co-crystal predictions of loratadine and desloratadine are listed.
| Compounds | Method | Success rate |
|---|---|---|
| Loratadine | CoForm | 32/41 = 78% |
| HBP | 31/41 = 76% | |
| MC | 16/41 = 39% | |
| Desloratadine | CoForm | 32/41 = 78% |
| HBP | 22/41 = 54% | |
| MC | 9/41 = 22% |
Conclusions
The potential for using co-crystallization technology to alter physical properties such as solubility and stability of various high-value organic solid-state materials is gaining traction. A key challenge is to be able to predict a priori which co-formers are most likely to produce new solid forms of the target compound. Therefore, we attempted to develop a fast and user-friendly software application to facilitate co-crystallization screening experiments. This app accurately predicted the outcome 78% for loratadine and 78% for desloratadine whereas HBP produced a success rate of 76% and 54% for loratadine and desloratadine, respectively. Finally, MC delivered a success rate of 39% and 22%, respectively.We hope this tool will be further tested, refined, and utilized by users interested in the crystalline solid-state, especially in the context of improving physical properties.26 In addition, the app is a customizable tool and will produce the most reliable outcomes, if the unknown target is a close match (similar molecular weight, rotatable bonds, functional groups) to the known targets in the database. Therefore, having a user-specific database will undoubtedly increase the predictive abilities of the app. We believe that the customizability of CoForm can extend its usability to hydrogen-bonded solids across areas such as pharmaceutics, agrochemicals, and energetic materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge the support from the Johnson Cancer Research Center at Kansas State University, and the Ann and Dave Braun Student Inventor Award. We also wish to thank Molly Vittengl supported by the National Science Foundation REU Site program under Grant numbers CHE-1852182 and CHE-1460898, and Lexi Berndt supported by U. S. Army Research Laboratory and the U. S. Army Research Office under contract/grant number W911NF-13-1-0387 for their contribution in the experimental work.References
- J. Berger, J. D. Dunn, M. M. Johnson, K. R. Karst and W. C. Shear, Am. J. Manag. Care, 2016, 22, S487–S495 Search PubMed
.
- C. Aakeröy, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2015, 71, 387–391 Search PubMed
- N. Bedeković, V. Stilinović and T. Piteša, Cryst. Growth Des., 2017, 17, 5732–5743 Search PubMed
- C. B. Aakeröy, T. K. Wijethunga, J. Benton and J. Desper, Chem. Commun., 2015, 51, 2425–2428 Search PubMed
- S. Li, T. Yu, Y. Tian, C. P. McCoy, D. S. Jones and G. P. Andrews, Mol. Pharmaceutics, 2016, 13, 3054–3068 Search PubMed
- D. D. Gadade and S. S. Pekamwar, Adv. Pharm. Bull., 2016, 6, 479–494 Search PubMed
- C. B. Aakeröy, S. Forbes and J. Desper, J. Am. Chem. Soc., 2009, 131, 17048–17049 Search PubMed
- S. Aitipamula, R. Banerjee, A. K. Bansal, K. Biradha, M. L. Cheney, A. R. Choudhury, G. R. Desiraju, A. G. Dikundwar, R. Dubey, N. Duggirala, P. P. Ghogale, S. Ghosh, P. K. Goswami, N. R. Goud, R. R. K. R. Jetti, P. Karpinski, P. Kaushik, D. Kumar, V. Kumar, B. Moulton, A. Mukherjee, G. Mukherjee, A. S. Myerson, V. Puri, A. Ramanan, T. Rajamannar, C. M. Reddy, N. Rodriguez-Hornedo, R. D. Rogers, T. N. G. Row, P. Sanphui, N. Shan, G. Shete, A. Singh, C. C. Sun, J. A. Swift, R. Thaimattam, T. S. Thakur, R. Kumar Thaper, S. P. Thomas, S. Tothadi, V. R. Vangala, N. Variankaval, P. Vishweshwar, D. R. Weyna and M. J. Zaworotko, Cryst. Growth Des., 2012, 12, 2147–2152 Search PubMed
- N. Sarkar, A. S. Sinha and C. B. Aakeröy, CrystEngComm, 2019, 21, 6048–6055 Search PubMed
- D. Musumeci, C. A. Hunter, R. Prohens, S. Scuderi and J. F. McCabe, Chem. Sci., 2011, 2, 883 Search PubMed
- J. McKenzie, N. Feeder and C. A. Hunter, CrystEngComm, 2016, 18, 394–397 Search PubMed
- N. Blagden, M. de Matas, P. T. Gavan and P. York, Adv. Drug Delivery Rev., 2007, 59, 617–630 Search PubMed
- M. A. Mohammad, A. Alhalaweh and S. P. Velaga, Int. J. Pharm., 2011, 407, 63–71 Search PubMed
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 Search PubMed
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 Search PubMed
- L. K. Mapp, S. J. Coles and S. Aitipamula, Cryst. Growth Des., 2017, 17, 163–174 Search PubMed
- P. A. Wood, N. Feeder, M. Furlow, P. T. A. Galek, C. R. Groom and E. Pidcock, CrystEngComm, 2014, 16, 5839 Search PubMed
- N. Sarkar and C. B. Aakeröy, Supramol. Chem., 2020, 32, 81–90 Search PubMed
- L. Fábián, Cryst. Growth Des., 2009, 9, 1436–1443 Search PubMed
- C. B. Aakeröy, N. C. Schultheiss, A. Rajbanshi, J. Desper and C. Moore, Cryst. Growth Des., 2009, 9, 432–441 Search PubMed
- C. B. Aakeröy, K. N. Epa, S. Forbes and J. Desper, CrystEngComm, 2013, 15, 5946 Search PubMed
- C. B. Aakeröy, D. J. Salmon, M. M. Smith and J. Desper, Cryst. Growth Des., 2006, 6, 1033–1042 Search PubMed
- C. B. Aakeröy, T. K. Wijethunga and J. Desper, New J. Chem., 2015, 39, 822–828 Search PubMed
- The Apache Groovy programming language, http://groovy-lang.org/, (accessed November 7, 2019)
- A. Delori, P. T. A. Galek, E. Pidcock, M. Patni and W. Jones, CrystEngComm, 2013, 15, 2916–2928 Search PubMed
- The software is available as a desktop application which can be downloaded and used with customized dataset tailored towards user-specific choice of co-formers .
Footnote |
| † Electronic supplementary information (ESI) available: HBP, MC, and CoForm prediction table and FT-IR grinding experiment data table. See DOI: 10.1039/d0ce01074j |
| This journal is © The Royal Society of Chemistry 2020 |