
Selective enclathration of xylenols: synergistic effects of mixed hosts†
Jacky S.
Bouanga Boudiombo
,
Hong
Su
 ,
Neil
Ravenscroft
,
Susan A.
Bourne
and
Luigi R.
Nassimbeni
*
,
Neil
Ravenscroft
,
Susan A.
Bourne
and
Luigi R.
Nassimbeni
*
Centre for Supramolecular Chemistry Research, Department of Chemistry, University of Cape Town, Rondebosch 7701, South Africa. E-mail: luigi.nassimbeni@uct.ac.za; Tel: +27 21 650 5893
First published on 22nd June 2020
Abstract
The six xylenol (XYL) isomers can be separated by selective enclathration with the host 4,4-isopropylidene bisphenol, H1. Crystal structures were elucidated for the following single and mixed guest inclusion compounds with H1: H1·34XYL (I), H1·35XYL (II), H1·23XYL·26XYL (III), H1·23XYL·35XYL (IV), where the xylenol isomers are abbreviated as, for example 34XYL for 3,4-xylenol. The crystal structures of selected H1·xylenols showed that there is extensive host⋯host and host⋯guest hydrogen bonding. Competition experiments with equimolar mixtures of pairs of xylenols (XYL) showed that the preference for inclusion was in the sequence 34XYL > 35XYL > 26XYL > 23XYL > 25XYL > 24XYL. By analogy to the Dutch resolution method (in which families of resolving agents are used to achieve chiral separations), two host compounds similar to H1 were used in pairs with H1 to improve the selectivity of the xylenols. 4,4′-(9-Fluorenylidene)bisphenol, H2, and 4,4′(cyclohexylidene)bisphenol, H3, were used in pairs with H1 and were shown to enhance the selectivity of a given xylenol which had been poorly separated by H1 alone. The crystal structure was elucidated for an unusual mixed host–mixed guest inclusion compound, H1·H2·26XYL/35XYL (V).
Introduction
The separation of mixtures of closely related molecules is an important chemical process and separation techniques are dependent on the difference in the physico-chemical properties of the individual components, and include several types of chromatography, crystallization, distillation, and evaporation. However, if the individual components have similar properties, standard techniques may not be suitable. This is the case with many isomers, while their separation remains important as they are frequently produced as mixtures in industrial processes. The individual components are usually more valuable than the mixture, because they form the feedstock for the syntheses of novel products.1Host–guest (or inclusion) chemistry has proved to be a useful methodology for the separation of closely related molecular species.2 When a host compound, H, is exposed to a mixture of guest molecules, A, B, C… this may result in an inclusion compound
| H + n1A + n2B + n3C… → H·Am |
The above represents an ideal case, in which A is exclusively selected. In practice this occurs seldom, and where there are many components a common strategy is to take the guests in pairs and analyse the final product by a suitable technique which yields its stoichiometry. By analysing the combination of all possible pairs of guests, it is possible to obtain the preferential affinity sequence of the host H for the complete series of guests A, B, C…
This methodology is driven by the phenomenon of molecular recognition, which is central to host–guest chemistry and crystal engineering. The special factors that lead to a suitable fit between host and guest molecules arise from the sum of multiple secondary interactions that impinge upon the molecular system. These secondary bonds are often directional, resulting in specificity and allowing a host molecule to discriminate between a given guest in a mixture of guests. This makes the host H selective, which is crucial to separation processes.
An important example of the separation of isomers is the petrochemical industry in which isomers of the C8 hydrocarbons (ortho-, meta-, para-, xylenes and ethyl benzene), the cresols and the xylenols are produced in large quantities from the catalytic reforming of crude oil. A well-known case is the separation of the isomers of xylene, which have similar normal boiling points ranging from 138.1 °C to 144.4 °C, rendering fractional distillation inefficient. This topic is the subject of a comprehensive review,3 which discusses various materials employed in the separation techniques that include metal–organic frameworks, zeolites and organic host molecules. Other contributions describe the optimal synthesis of p-xylene separation4 and the use of Werner clathrates for the separation of xylenes from in the vapour state and the concomitant kinetics of adsorption.5
Hydrocarbons are not the only compounds that have been separated by enclathration and there are several recent examples where mixtures of both aliphatic and aromatic compounds have been selectively included into host–guest complexes.6–10
A particularly challenging problem for the separation of isomers is that of enantiomer resolution of a racemic modification. Here, the two enantiomeric components have identical physical properties such as melting point, boiling points, refractive index, density, dipole moment, and only differ in their response to polarized light. One successful strategy for separating enantiomers combines a chiral resolving agent which forms a compound preferentially with either one or the other enantiomer. This separation process is not always routine and may yield incomplete resolution. However, a significant advancement was made by the discovery by T. Vries et al.11 who used a combinational approach of related “families” of resolving agents to improve the resolution of racemates. This has been summarized in the Handbook of Optical Resolutions edited by D. Kozma12 and is known as the “Dutch Resolution Method”. We were inspired by this idea to extend the usual host–guest method of separation of isomers to an analogy of the Dutch resolution method, in which we employed pairs of similar host compounds for the separation of isomers from binary mixtures, with the aim of obtaining enhanced selectivity of the guest species.
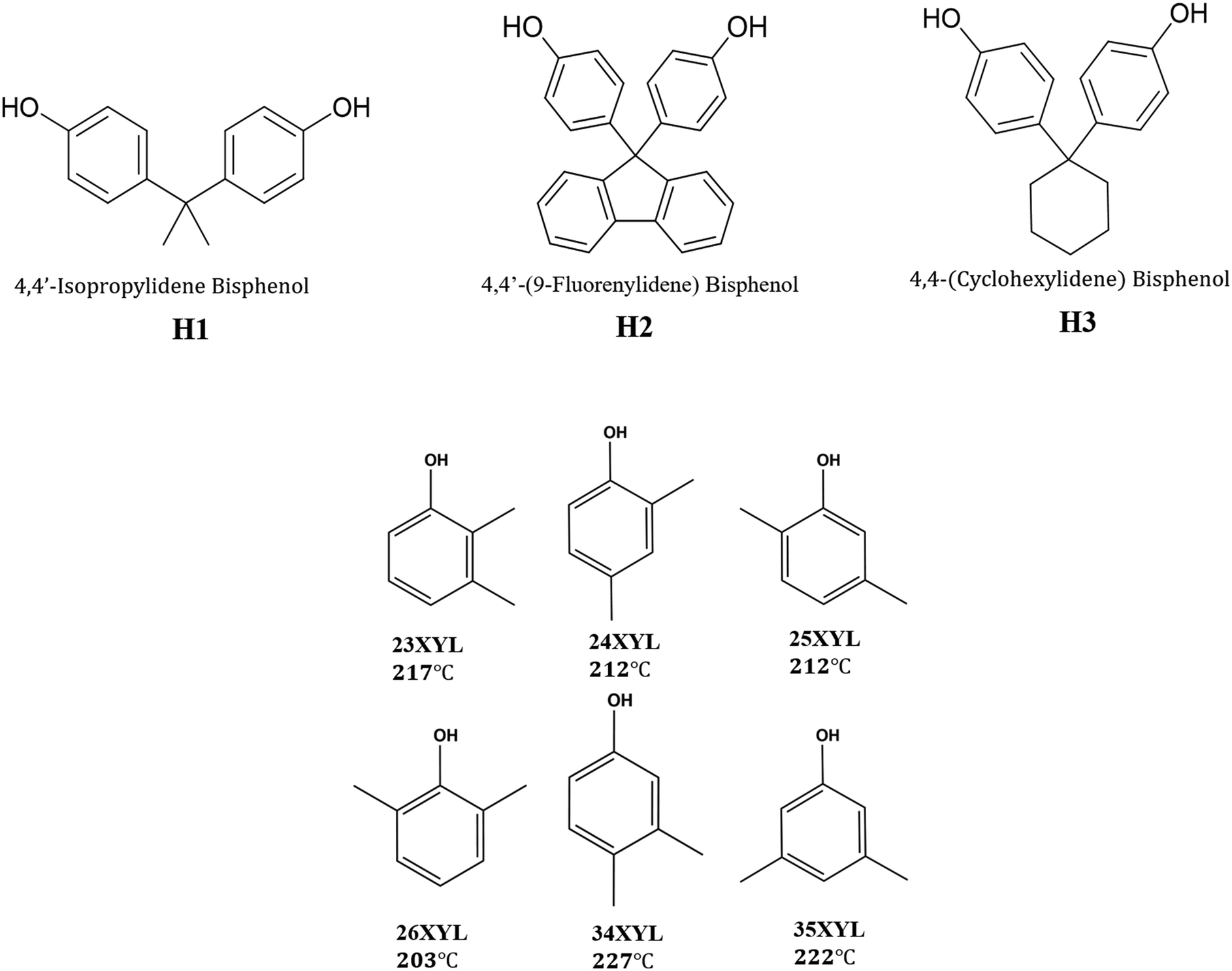
In this work, we aimed to achieve separation of the six xylenol isomers, by selective inclusion using three organic host compounds which contain the common bisphenol moiety. The structures of the host compounds are shown in Scheme 1, which also presents the six xylenol isomers with their normal boiling points, as well as the abbreviations used for these compounds.
 | ||
| Scheme 1 | ||
Experimental
Materials
The compounds were acquired from Sigma-Aldrich and were used without further purification.Competition experiments
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :host was thus 10:1, to ensure that an excess of both guests is always available to the host, thus eliminating an artificial selectivity developing as compounds crystallize from the solution. Ethyl acetate was chosen as the common solvent to overcome any possible solubility barriers and because it is not included by any of the host compounds, so would not be incorporated in the crystalline products. Details of the resulting crystals are given in Tables 1 and 2 in the Results section. Inclusion compounds I (H1·34XYL), II (H1·35XYL), III (H1·23XYL/26XYL), and IV (H1·23XYL/35XYL) were selected from these experiments and subjected to single crystal diffraction as detailed below.
:host was thus 10:1, to ensure that an excess of both guests is always available to the host, thus eliminating an artificial selectivity developing as compounds crystallize from the solution. Ethyl acetate was chosen as the common solvent to overcome any possible solubility barriers and because it is not included by any of the host compounds, so would not be incorporated in the crystalline products. Details of the resulting crystals are given in Tables 1 and 2 in the Results section. Inclusion compounds I (H1·34XYL), II (H1·35XYL), III (H1·23XYL/26XYL), and IV (H1·23XYL/35XYL) were selected from these experiments and subjected to single crystal diffraction as detailed below.
| Xylenol (XYL) | 23XYL | 24XYL | 25XYL | 26XYL | 34XYL | 35XYL |
|---|---|---|---|---|---|---|
| 24XYL | [1] | |||||
| 23XYL-90 | ||||||
| 24XYL-10 | ||||||
| 25XYL | [2] | [3] | ||||
| 23XYL-55 | Host | |||||
| 25XYL-45 | ||||||
| 26XYL | [4] | [5] | [6] | |||
| 23XYL-40 | Host | 25XYL-6 | ||||
| 26XYL-60 | 26XYL-94 | |||||
| 34XYL | [7] | [8] | [9] | [10] | ||
| 23XYL-10 | 24XYL-16 | 25XYL-6 | 26XYL-3 | |||
| 34XYL-90 | 34XYL-84 | 34XYL-94 | 34XYL-97 | |||
| 35XYL | [11] | [12] | [13] | [14] | [15] | |
| 23XYL-46 | 24XYL-9 | 25XYL-7 | 26XYL-12 | 34XYL-94 | ||
| 35XYL-54 | 35XYL-91 | 35XYL-93 | 35XYL-88 | 35XYL-6 |
| Structure | I | II | III | IV |
|---|---|---|---|---|
| Compound | H1·34XYL | H1·35XYL | H1·23XYL/26XYL | H1·23XYL/35XYL |
| Formula asymm. unit | (C15H16O2)·C8H10O | (C15H16O2)·C8H10O | 2(C15H16O2)·2C8H10O | 4(C15H16O2)·4C8H10O |
| M [g mol−1] | 350.4 | 350.4 | 700.9 | 1402 |
| Data collection temp T [K] | 173(2) | 173(2) | 173(2) | 173(2) |
| Crystal shape and size [mm] | Orange block, 0.23 × 0.28 × 0.30 | Orange block, 0.05 × 0.06 × 0.10 | Orange block, 0.18 × 0.25 × 0.28 | Colourless needle, 0.05 × 0.09 × 0.48 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c | C2/c | Pn |
| a [Å] | 6.4218(6) | 11.783(3) | 30.650(2) | 6.3007(3) |
| b [Å] | 14.8289(14) | 11.175(2) | 6.3048(5) | 20.7627(11) |
| c [Å] | 20.2173(19) | 15.163(3) | 39.852(3) | 29.9858(14) |
| β [°] | 96.132(2) | 93.080(4) | 91.359(2) | 95.787(2) |
| Volume [Å3] | 1914.2(3) | 1993.8(8) | 7699(1) | 3902.7(3) |
| Z | 4 | 4 | 8 | 2 |
| D c, calc. density [g cm−3] | 1.216 | 1.209 | 1.193 | |
| Absorption coefficient [mm−1] | 0.079 | 0.079 | 0.078 | |
| F(000) | 752 | 3008 | 1504 | |
| θ range | 1.706–28.339 | 1.329–27.910 | 1.962–26.385 | |
| Reflections collected | 30151 |
101365 |
69771 |
|
| No. independent reflections | 4783 | 9208 | 14770 |
|
| No. reflections with l > 2sigma(I) | 3382 | 8537 | 14075 |
|
| R int | 0.0654 | 0.0255 | 0.0395 | |
| Final R indices, R1, wR2 [I > 2sigma(I)] | 0.0486, 0.1095 | 0.0387, 0.0996 | 0.0412, 0.1009 | |
| R indices (all data), R1, wR2 | 0.0753, 0.1226 | 0.0417, 0.1021 | 0.0440, 0.1025 | |
| Max, min residual electron density (e Å−3) | 0.220, −0.225 | 0.309, −0.175 | 0.263, −0.188 |
:(total) hosts was thus 10:1. The solution was allowed to crystallize as before. Compound V (H1·H2·26XYL/35XYL) crystallized from the mixture of the two hosts with 2,6-xylenol and 3,5-xylenol, and was subjected to single crystal diffraction as described below.
:(total) hosts was again 10:1. The solutions were allowed to crystallize as before.
The crystals were harvested and blotted dry and subjected to NMR and single crystal X-ray diffraction analysis. The crystals were not washed with a different solvent for fear of partial dissolution and loss of the included xylenol guests.
X-ray crystallography
Single crystal X-ray diffraction data were collected on a Bruker DUO APEX II diffractometer for all structures using Mo Kα radiation (λ = 0.71073 Å) at a temperature of 173 K.13 The intensity data were collected using the phi scan and omega scan techniques, scaled, and reduced with SAINT-Plus.14 The correction of the collected intensities for absorption was done using the SADABS program.15 The X-seed interface,16 operating the SHELX suite of programs17 was used to solve each structure by direct methods, and to carry out structure refinement using full-matrix least squares. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms bound to carbon were included in the refinement in idealized positions in a riding model with isotropic thermal parameters 1.2–1.5 times the Uiso values of their parent atoms. Hydrogens on the hydroxyl groups of both the hosts and guests were placed according to the formulae devised by Lusi and Barbour,18 who analyzed the data of H-bonded system obtained from neutron diffraction. The method offers an equation based on the O(donor)⋯O(acceptor) distance which yields the value of the O–H bond length in H-bonded O–H⋯O interactions.NMR spectroscopy
Approximately 5 mg of representative crystals were blotted dry, crushed, and dissolved in 600 μL of DMSO-d6 and introduced into a 5 mm NMR tube for data acquisition. One-dimensional (1D) 1H and two-dimensional (2D) 1H–13C HSQC NMR spectra were recorded on a Bruker 300 or 400 MHz spectrometer at 30 °C and processed using standard Bruker software (Topspin 3.5). The HSQC experiment was optimized for J = 145 Hz (for directly attached 1H–13C correlations). The spectra were referenced relative to the solvent signal at 7.26 ppm (for 1H) and 77.16 ppm (for 13C); appropriate signals were integrated to determine the relative proportions of the guests. The analysis was carried out on the crystals harvested from the mother liquors of the equimolar xylenol binary mixtures. These crystals were dried using blotting paper but were not washed with any solvent to avoid any exchange of guests.Integration of the peaks of the methyl substituents of the isomers were used to determine their relative proportions of guests in the different samples. Fig. 1 shows an overlay of the expansion of the methyl region of the 1H NMR spectra of the guests (23XYL and 26XYL) and the host and guests. Fig. 1a shows the methyl groups of the host compound at 1.15 ppm. Fig. 1b has CH3-2 at 2.03 ppm and CH3-3 at 2.18 ppm for 23XYL and Fig. 1c has CH3-2 and CH3-6 at 2.15 ppm. These diagnostic peaks were used to determine the relative ratio of 60/40% for the 23XYL/26XYL guests in the host–guests mixture (Fig. 1d). The same procedure was applied to all other mixtures and gave the results shown in Table 1. Representative NMR spectra are included in the ESI.† When two isomers had overlapping methyl peaks in the 1H spectrum, but the 13C NMR signals were resolved, then quantification was performed by integration of the relevant HSQC cross peaks. Thus, NMR analysis elucidated the relative ratio of the guests, whereas the percentage host–host and host–guest was also obtained when mixed hosts method was used.
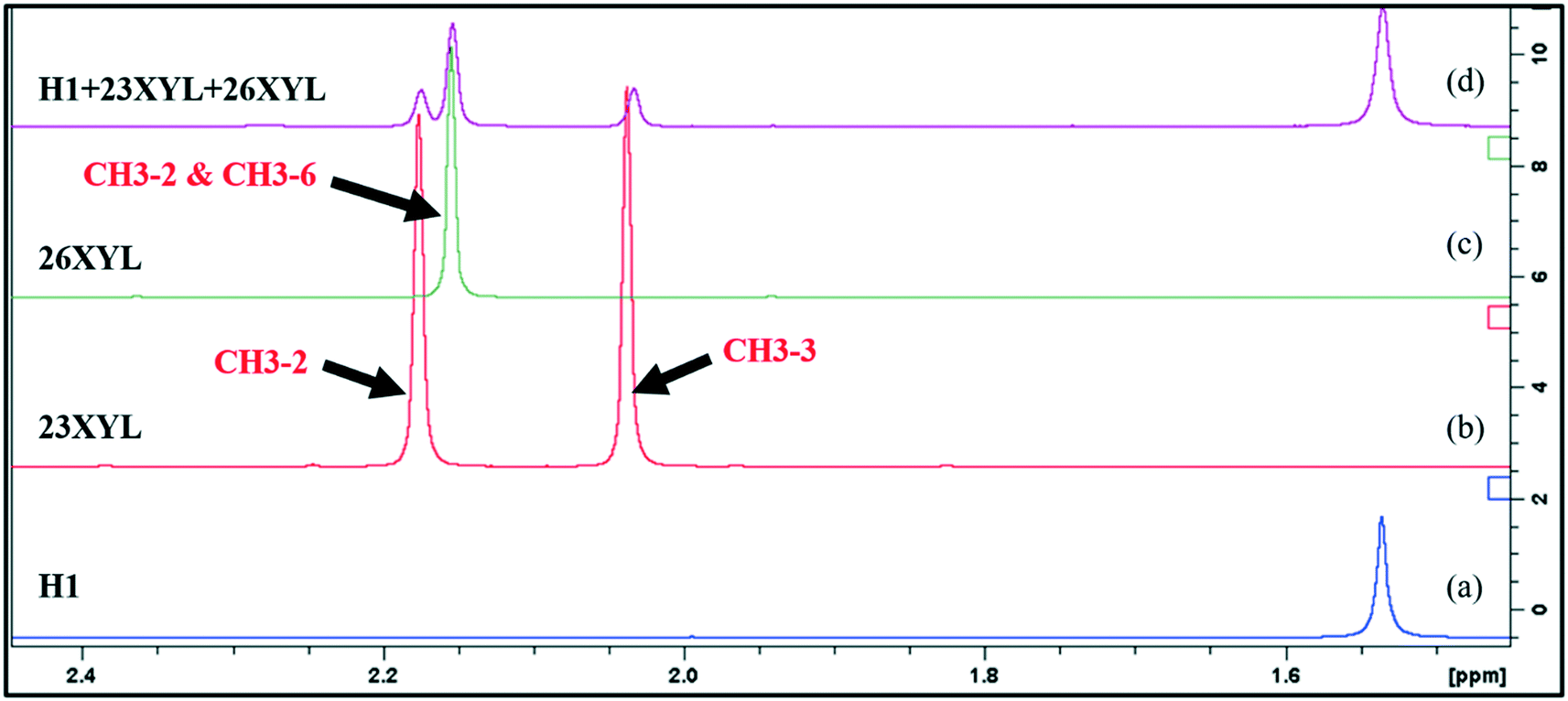 | ||
| Fig. 1 Overlay of the expansion of the 1H NMR methyl region for (a) H1, (b) 23XYL, (c) 26XYL, (d) complex of H1 + 23XYL + 26XYL obtained in experiment [4]. | ||
Results
Competition experiments: separation of xylenols by H1
To study the selectivity of the host H1 for particular isomers of xylenol, we carried out pairwise competition experiments in which H1 was dissolved with an equimolar mixture of two xylenol isomers and the resulting crystals analysed by NMR and, where possible, by single crystal structure elucidation. Table 1 shows the NMR results given as percentages of the enclathrated xylenol guests. In general, when both xylenols were enclathrated, if the major component was greater than 85%, single crystal analysis found that only the major component can be refined in the crystal structure. We have observed a similar phenomenon in the separation of lutidine isomers.19–21 The most interesting result is obtained when the selectivity is poor because, paradoxically, more information is obtained regarding the selectivity when both isomers are entrapped in the same crystal. The resultant conformational changes in the host can thus be studied and one may be able to examine the mechanism giving rise to selectivity.Experiments [3] and [5] resulted in the recrystallisation of the empty host structure (sometimes referred to as the apohost structure). Although experiments [1] and [6] indicated high selectivity for 23XYL and 26XYL respectively, they yielded poor quality crystals which could not be used for data collection. Experiments [7], [8], [9] [10] and [15] showed a high selectivity for 34XYL, and a crystal was selected from [15] for X-ray analysis (crystal structure I). 35XYL appeared to be selected in experiments [12], [13] and [14] and inclusion compound II was selected for crystal structure analysis from the latter. Experiments [4] and [11] resulted in the crystallization of mixed guest inclusion compounds III and IV respectively. Experiment [2] also showed evidence of a mixed-guest compound, but the crystals did not diffract adequately for single crystal analysis. In total, we elucidated four crystal structures from the competition experiments given in Table 1. Their crystal data and refinement parameters are listed in Table 2. The identity of the guests included can be unequivocally confirmed for those cases where single crystal diffraction was performed, but we note that similar confirmation is not possible for the experiments where single crystal structures were not obtained. However, we saw no evidence, on inspection under polarized light, of two or more crystalline phases being produced in any of these or subsequent experiments.
Crystal structures from competition experiments with host H1
The crystal structures exhibit networks of hydrogen bonds in which the hydroxyl moieties of the host H1 and the xylenol guests can act as both H-bond donor and acceptor.Structure I, H1·34XYL, crystallizes in P21/c with Z = 4. The packing is characterized by chains of H1 which are stabilized by O–H⋯O(H) H-bonds, and in addition, form H-bonded rings with the 34XYL guest. The packing is shown in Fig. 2, and may be described by graph-set analysis22,23 as C11(12)[R33(6)]. The H1 chains propagate in the [010] direction and the 34XYL guests are in the loops of the twisted chains.
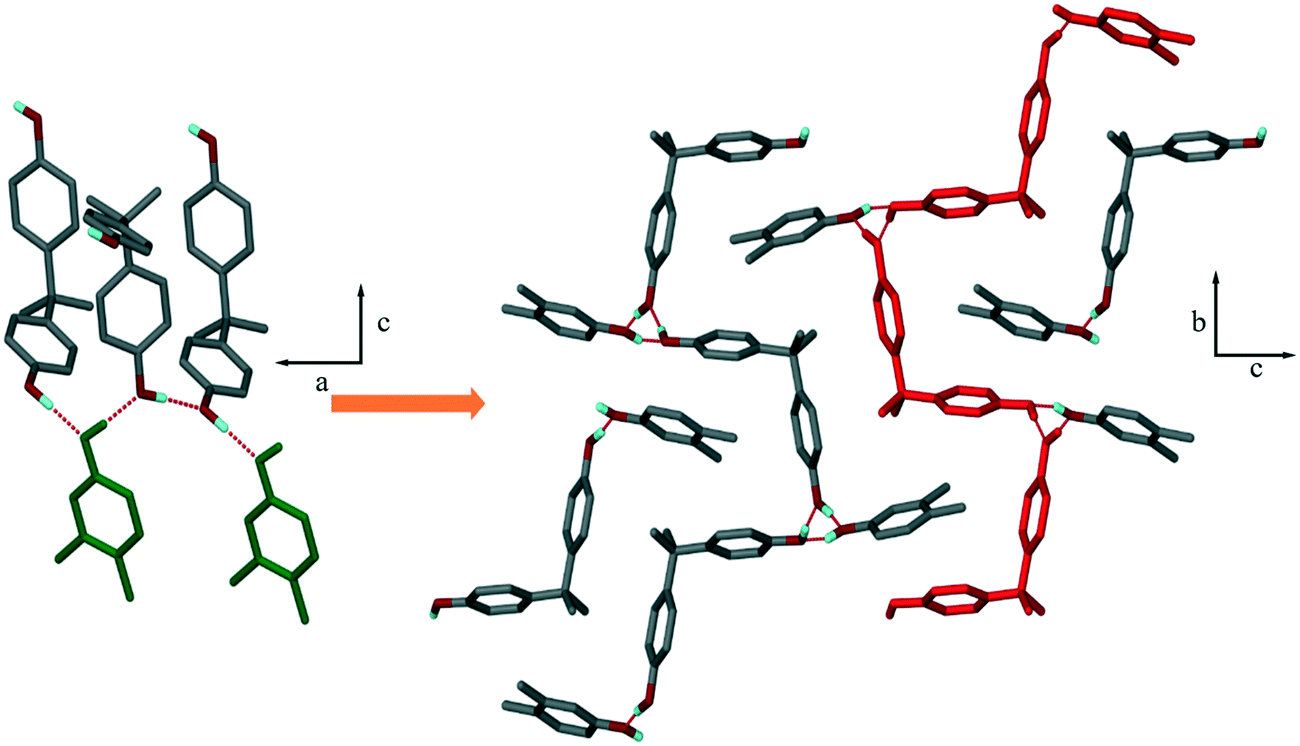 | ||
| Fig. 2 Packing of compound I (H1·34XYL) viewed along [010] (left) and along [100] (right). Alternative host chains in red (atom colours are C: grey, O: red, H: blue) hydrogens bound to carbon are omitted for clarity. | ||
A search of the Cambridge Structural Database24 found 11 structures in which H1 had formed a co-crystal or solvate. One of these, refcode SIXDOS, is the co-crystal of H1 with p-cresol.25 This compound is isostructural with compound I, having almost identical unit cell parameters and packing arrangements. Although I has an extra methyl group at the meta-position, the same hydrogen-bonding motifs can be formed in both structures, which accounts for this similarity. The CSD search revealed 20 solvate or co-crystal structures with H2 and 47 with H3, though none are similar to the structures reported here.
Structure II, H1·35XYL could not be refined satisfactorily and we only report the cell parameters in Table 2. However, the structure could be solved sufficiently to show that it bears certain packing similarities with I, in that the host forms a series of chains running along [010], with heavily disordered 35XYL guests forming hydrogen bonded bridges between chains (Fig. 3).
 | ||
| Fig. 3 Packing of compound II viewed along [100]. Colour coding as in Fig. 2. | ||
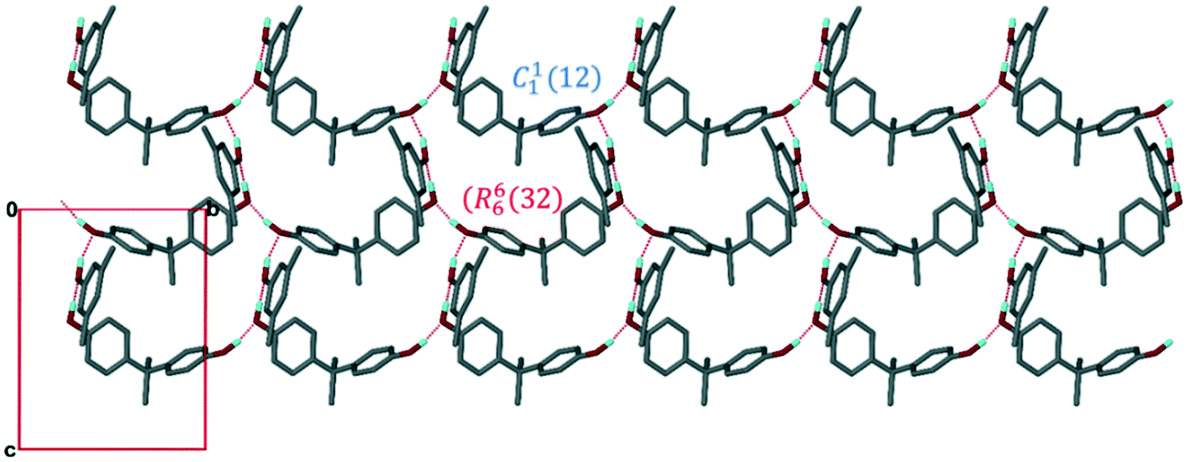
Structure III crystallizes in the space group C2/c. The asymmetric unit consists of two H1 hosts, a disordered 23XYL (86% site occupancy) and 26XYL (14% site occupancy) and one 26XYL with full site occupancy. The ratio of H1:23XYL:26XYL is thus 1:0.43:0.57 which is in good agreement with the NMR data in Table 1 for experiment [4]. The packing, shown in Fig. 4 shows the H1 hosts packed in double layers running along [001]. The interior of the double layer is hydrophobic, containing the gem-dimethyl groups, while the outer sides of the double layer may be deemed hydrophilic in that it contains the hydroxyl moieties which hydrogen bond with the 23XYL and 26XYL guests in a series of R33(6) hydrogen bonded rings.
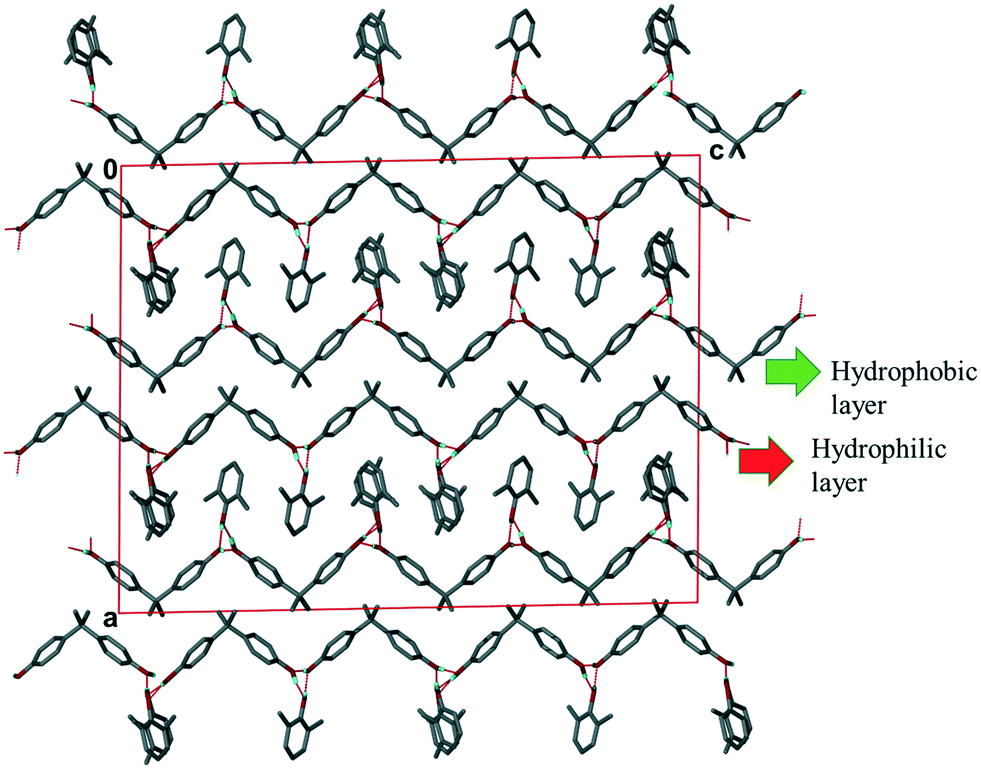 | ||
| Fig. 4 Packing of III (H1·23XYL/26XYL) viewed along [100] showing the two layers formed within the structure. | ||
The refinement of structure IV (H1·23XYL/35XYL) proved difficult. Although the synthesis was repeated several times, the resultant crystals were of poor quality. The best preparation yielded crystals that did not extinguish completely under polarized light and the structure was initially solved in the space group P1. The resultant structure was checked for higher symmetry by the program Platon26 which strongly suggested the space group Pn, duly adopted. Platon further identified twinning which was resolved by application of the appropriate twin law.
The asymmetric unit contains four H1 hosts, two 23XYL and two 35XYL guest, all crystallographically independent. The ratio of H1:23XYL:35XYL is thus 1:0.50:0.50, which is in good agreement with the ratio 1:0.46:0.54 determined by NMR on the bulk sample (Table 1, experiment [11]). The packing, shown in Fig. 5, bears strong resemblances to that shown for structure III, in that one notes a host double layer with hydrophobic and hydrophilic surfaces. The latter features H-bonded 23XYL and 35XYL guests. The hosts and guests form chains running along [010].
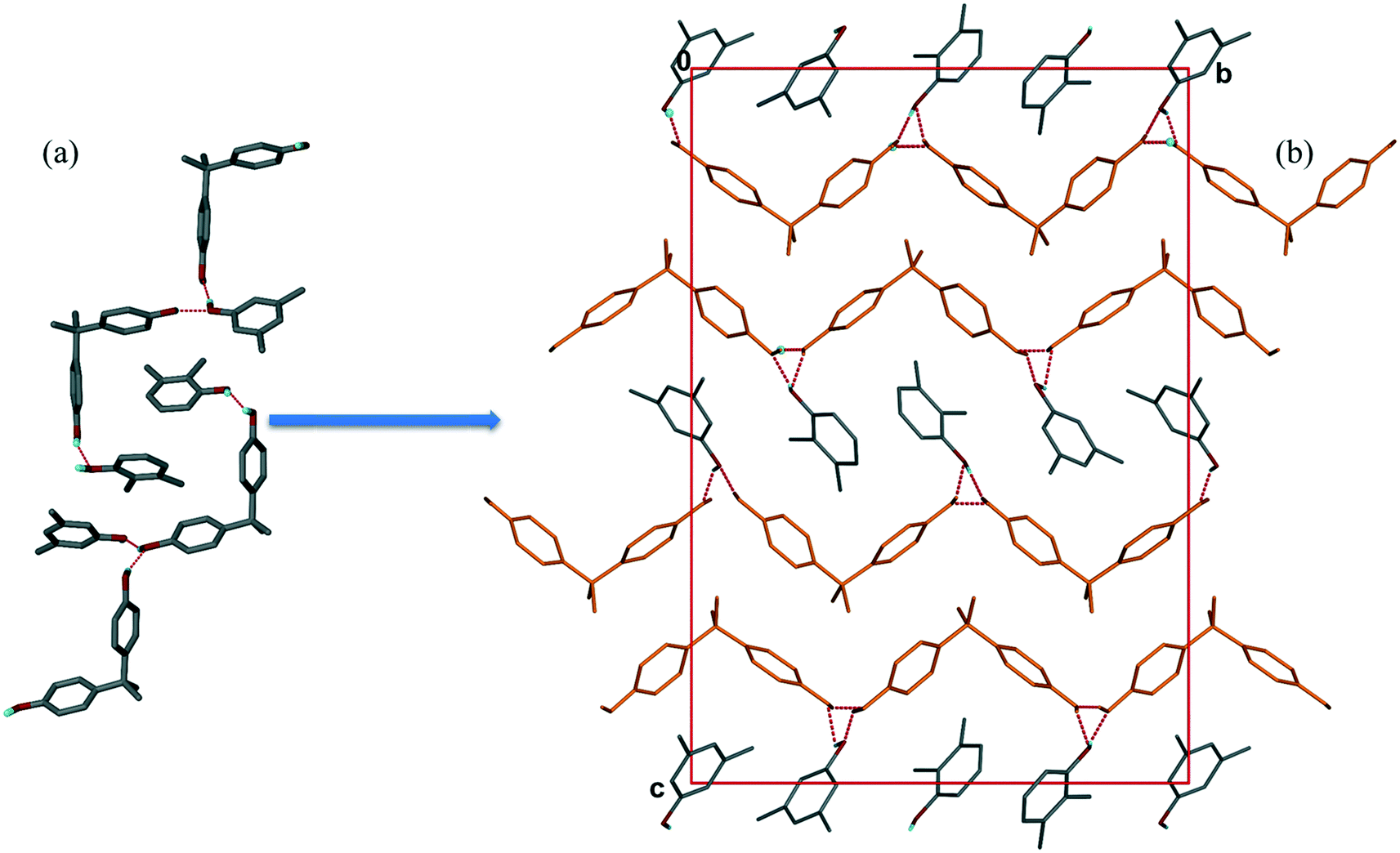 | ||
| Fig. 5 Packing of IV showing (a) hydrogen bonding between host and host, and host and guest, (b) host forms H-bonded chains (in orange) parallel to the b-axis. | ||
The conformation of the host H1 in all the structures elucidated is reported in Table S1† which lists the torsion angles τ1 and τ2 are defined as τ1 = (a − b − c − d) and τ2 = (b − c − d − e), Scheme 2. The variations of the torsion angles τ1 and τ2 are relatively small, showing that the host conformation is fairly constant and is thus does not play a key role in the selectivity of xylenol isomers.
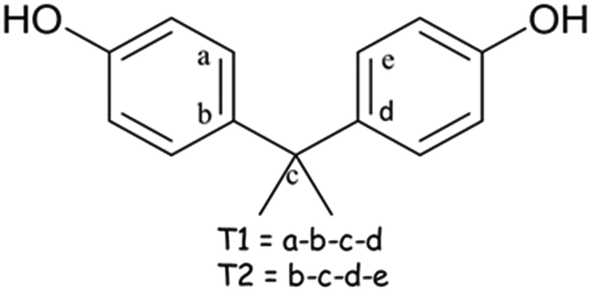 | ||
| Scheme 2 Torsion angles of H1. | ||
Competition experiments: separation of xylenols by mixed hosts (H1 and H2)
The Dutch resolution method11 was first reported in 1998, and involves the use of combinations of structurally similar resolving agents (“families”) to enhance the selective resolution of enantiomers. We hypothesized that the use of combinations of structurally similar host compounds might enhance the selectivity for isomers compared to the use of a single host compound. To that end, we carried out competition experiments using a combination of two hosts (either H1 and H2, or H1 and H3) across the pairwise combinations of xylenol isomers. As before, the competition experiments were initiated with a starting (total) guests:(total) hosts ratio of 10:1 to eliminate an artificial selectivity developing as compounds crystallize from the solution.
Experiments [16], [17], and [18] tested the selectivity H2 alone in competition experiments of 23XYL/26XYL, 23XYL/35XYL, and 26XYL/35XYL. These were chosen because H1 had shown poor selectivity in the first two combinations while the third combination yielded a strong preference for 35XYL (Table 1). The results testing the efficacy of H2 are reported in Table 3, and show a similar lack of selectivity for 23XYL/26XYL, a preference for 35XYL in 26XYL/35XYL and an inversion in selectivity, towards 23XYL in 23XYL/35XYL.
| Composition | H1 | H2 | 23XYL | 26XYL | 35XYL |
|---|---|---|---|---|---|
| Expt [16] start | — | 100 | 50 | 50 | — |
| End | — | 100 | 50 | 50 | — |
| Expt [17] start | — | 100 | 50 | — | 50 |
| End | — | 100 | 81 | — | 19 |
| Expt [18] start | — | 100 | — | 50 | 50 |
| End | — | 100 | — | 28 | 72 |
| Expt [19] start | 50 | 50 | 50 | 50 | — |
| End | 98 | 2 | 55 | 45 | — |
| Expt [20] start | 50 | 50 | 50 | — | 50 |
| End | 48 | 52 | 88 | — | 12 |
| Expt [21] start | 50 | 50 | — | 50 | 50 |
| End | 51 | 49 | — | 38 | 62 |
Experiments [19]–[21] detail the results of the mixed host/mixed guest competition experiments with H1 and H2. An interesting aspect of the Dutch resolution method is that the final crystalline product may have host–host and host–guest stoichiometries that differ from those of the starting solution. This was particularly evident in experiment [19] where an equimolar mixture of H1 and H2 resulted in crystals containing almost entirely H1, while the guest selectivity was poor in this case. Experiments [20] and [21] retained the H1:H2 ratio and showed altered selectivity towards the xylenols compared to single host experiments. In experiment [20], the competition with 23XYL/35XYL, the selectivity changes from 54% 35XYL (with H1), or 81% 23XYL (with H2) to 88% 23XYL in the mixed host system. In experiment [21], the competition with 26XYL/35XYL, the selectivity changes from 88% 35XYL (with H1), or 72% 35XYL (with H2) to 62% 35XYL in the mixed host system. Understanding these results would be enhanced if one were able to obtain single crystals of each of these outcomes. Unfortunately, this is often not possible, but we were able to obtain a crystal structure from the product of experiment [21].
Structure V is unusual in that it was derived from a solution containing an equimolar mixture of H1 and H2, and an equimolar mixture of 26XYL and 35XYL, and retained all four species in the crystalline product. The NMR results showed that the bulk sample contained an almost equimolar amount of H1 and H2, which had enclathrated an unequal mixture of 26XYL and 35XYL. The resulting product crystallized in P21/c and the asymmetric unit contains two H1 molecules, two H2 molecules and a disordered 26XYL/35XYL sharing the same site in unequal proportions (26XYL:35XYL = 0.34:0.66, in good agreement with NMR data). The important feature of this structure is that the disordered 26XYL/35XYL guest is surrounded by four host molecules (one pair of H1 and one pair of H2) as shown in Fig. 6. The crystallographic data parameters of V are given in Table 4. Mixed-host, mixed-guest crystal structures are still relatively rarely reported in the literature. One of us has previously reported a similar example, in which a family of four similar diol hosts were used singly and in pairs in an attempt to enhance the resolution of 2-butylamine.27 Although there was no improvement in the enantiomeric resolution, a structure of two hosts with two guests was reported.
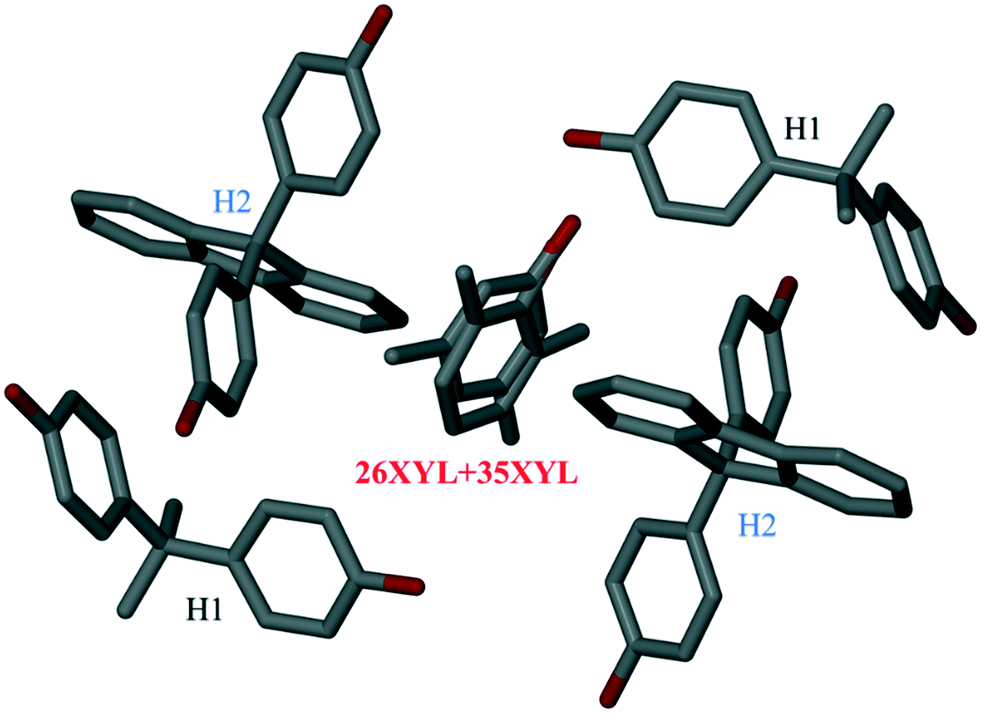 | ||
| Fig. 6 Asymmetric unit of structure V showing 26XYL/35XYL enclathrated by a mixture of the hosts H1 and H2 (atom colours as per Fig. 2. Hydrogen atoms have been omitted for clarity). | ||
| V | |
|---|---|
| Compound | H1·H2·26XYL/35XYL |
| Formula asymm. unit | 2(C15H16O2)2(C25H18O2)·C8H10O |
| M [g mol−1] | 1279.5 |
| Data collection temp T [K] | 173(2) |
| Crystal shape and size [mm] | Orange block, 0.18 × 0.20 × 0.32 |
| Crystal system | Monoclinic |
| Space group | P21/c |
| a [Å] | 21.0580(9) |
| b [Å] | 15.5380(6) |
| c [Å] | 21.249(1) |
| β [°] | 98.481(1) |
| Volume [Å3] | 6876.6(5) |
| Z | 4 |
| D c, calc. density [g cm−3] | 1.236 |
| Absorption coefficient [mm−1] | 0.079 |
| F(000) | 2712 |
| θ range | 0.978–28.370 |
| Reflections collected | 78563 |
| No. independent reflections | 17204 |
| No. reflections with l > 2sigma(I) | 11140 |
| R int | 0.0707 |
| Final R indices, R1, wR2 [I > 2sigma(I)] | 0.0500, 0.1110 |
| R indices (all data), R1, wR2 | 0.0894, 0.1300 |
| Max, min residual electron density (e Å−3) | 0.239, −0.208 |
Competition experiments: separation of xylenols by mixed hosts (H1 and H3)
The selectivity of the same three pairs of isomers was studied with combinations of H1 and H3. The latter host, 4,4-cyclohexylidene bisphenol has been studied extensively and has been shown to enclathrate picolines,28 aliphatic alcohols29 and lutidines.30 It has also been employed in the separation of xylenols,31 establishing that 35XYL is preferred to 26XYL; 23XYL is preferred to 26XYL; but the competition between 35XYL and 23XYL is concentration dependent. In this work, equimolar mixtures of H1and H3 with equimolar mixtures of the xylenols gave rise to products in which both host and guest ratios had changed, with an apparent selectivity for H3 over H1 and for 23XYL over 26XYL, and 35XYL in both experiments where it was present (Tables 5 and S4 and S5†).| Composition | H1 | H3 | 23XYL | 26XYL |
|---|---|---|---|---|
| Expt [22] start | 90 | 10 | 50 | 50 |
| End | 100 | 0 | 55 | 45 |
| Expt [23] start | 80 | 20 | 50 | 50 |
| End | 73 | 27 | 65 | 35 |
| Expt [24] start | 70 | 30 | 50 | 50 |
| End | 38 | 62 | 72 | 28 |
| Expt [25] start | 60 | 40 | 50 | 50 |
| End | 9 | 91 | 90 | 10 |
| Expt [26] start | 50 | 50 | 50 | 50 |
| End | 6 | 94 | 92 | 8 |
The observation that the relative amounts of both host and guest vary in this way led us to explore the phenomenon further by varying the starting ratio of H1:H3 systematically for each pairwise combination of xylenol isomers. The results for the H1/H3 and the guest pair 23XYL/26XYL are reported in Table 5 and shown graphically in Fig. 7. The results for the other isomer pairs are given in the ESI† as Tables S4 and S5.
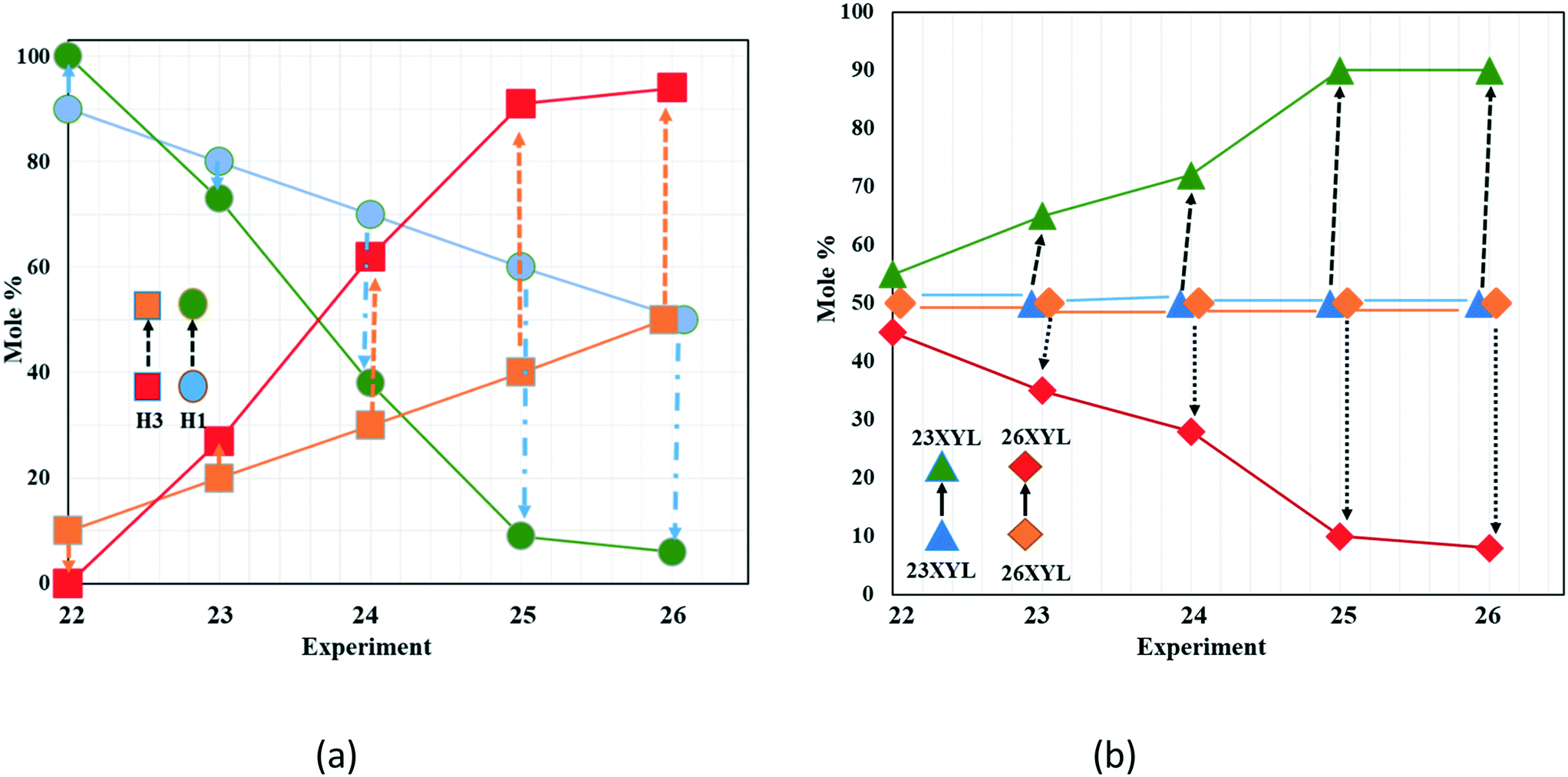 | ||
| Fig. 7 Trends observed in the competition experiments varying both host and guest ratios. (a) Host H1 decreases while H3 is selected; (b) guest 23XYL is consistently selected across all compositions. | ||
Fig. 7a shows H1  starting at 90% and decreasing linearly to 50%, while H3
starting at 90% and decreasing linearly to 50%, while H3  starts at 10% and increases to 50%. The trend of the experiments shows H1
starts at 10% and increases to 50%. The trend of the experiments shows H1  has decreased at the expense of H3
has decreased at the expense of H3  in the products obtained. The concomitant selectivity of the 23XYL/26XYL (Fig. 7b) shows a steady increase in 23XYL and decrease in 26XYL. There is thus a direct correlation between H1/H3 and the corresponding selectivity of 23XYL/26XYL. Similar results are obtained for H1/H3 and the guests 23XYL/35XYL (Table S4, Fig. S1†). However, Table S5† shows that changes in the H1/H3 composition had no effect in the 26XYL/35XYL selectivity, with 35XYL being exclusively enclathrated.
in the products obtained. The concomitant selectivity of the 23XYL/26XYL (Fig. 7b) shows a steady increase in 23XYL and decrease in 26XYL. There is thus a direct correlation between H1/H3 and the corresponding selectivity of 23XYL/26XYL. Similar results are obtained for H1/H3 and the guests 23XYL/35XYL (Table S4, Fig. S1†). However, Table S5† shows that changes in the H1/H3 composition had no effect in the 26XYL/35XYL selectivity, with 35XYL being exclusively enclathrated.
Conclusion
The separation of the six xylenol isomers has been carried out by enclathration employing 4,4-isopropylidene bisphenol, H1, with a selectivity preference of 34XYL > 35XYL > 26XYL > 23XYL > 25XYL > 24XYL. Crystal structure analysis of two single-guest and two mixed-guest structures showed that the host conformation remains fairly constant, thus eliminating this as the driving factor in the selectivity observed. Applying a modification of the Dutch resolution method, in which a family of similar hosts are combined, gave rise to greater guest selectivity. The combination of H1 with H2 in equimolar proportions enhances 23XYL over 35XYL, and 35XYL over 26XYL. The latter combination yielded a structure (V) comprising both H1 and H2, and guest refinement showed a selectivity in good agreement with that analysed in the bulk sample by NMR. The combination of H1 with H3 was carried out in different experiments which altered the proportion of H1/H3 systematically. Enclathration increased the proportion of H3 and enhanced the selectivity of 23XYL over 26XYL.The crystal structures I, II, III, and IV obtained with H1 are all stabilized by extensive hydrogen bonded networks, linking adjacent host molecules into chains which also hydrogen bonded the captured guests. The strength of the hydrogen bonds, as estimated by the O(donor)⋯O(acceptor) distances vary from 2.64 Å to 2.95 Å, and may be considered to change from strong to weak.32 These may be regarded as a constant feature throughout the structures elucidated. The synergistic selectivity effects which occur with the addition of a second host molecule may be attributed to the packing effects brought about by the different moieties (fluorenylidene in H2 and cyclohexylidene in H3). Confirmation of this effect will be sought in further work aiming to isolate crystals of these mixed-host, mixed-guest compounds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr. Clive Oliver (University of Cape Town) for useful discussions regarding the crystal structures.References
- R. S. Ward and A. Pelter, J. Chromatogr. Sci., 1974, 12, 570–574 CrossRef CAS
.
- L. R. Nassimbeni, Supramol. Chem., 2000, 12, 161–167 CrossRef CAS
- Y. Yuxi, B. Peng and G. Xianghai, Ind. Eng. Chem. Res., 2017, 56, 14727–14753 Search PubMed
- R. M. Lima and I. E. Grossman, AIChE J., 2009, 55, 354–373 CrossRef CAS
- M. Lusi and L. J. Barbour, Angew. Chem., Int. Ed., 2012, 51, 3928–3931 CrossRef CAS PubMed
- J. S. Bouanga Boudiombo, H. Su, S. A. Bourne and L. R. Nassimbeni, Cryst. Growth Des., 2018, 18, 424–430 CrossRef CAS
- E. J. Tiffin, N. M. Sykes, E. Weber, N. Ravenscroft and L. R. Nassimbeni, Cryst. Growth Des., 2019, 19, 1880–1887 CrossRef CAS
- B. Barton, L. de Jager, U. Senekal and E. C. Hosten, J. Inclusion Phenom. Macrocyclic Chem., 2019, 95, 259–271 CrossRef CAS
- B. Barton, U. Senekal and E. Hosten, Tetrahedron, 2019, 75, 3399–3412 CrossRef CAS
- B. Barton, L. de Jager and E. C. Hosten, CrystEngComm, 2019, 21, 4387–4400 RSC
- T. Vries, H. Wynberg, E. van Echten, J. Koek, W. ten Hoeve, R. M. Kellogg, Q. B. Broxterman, A. Minnard, B. Kaptein, S. van der Sluis, L. Hulshof and J. Kooistra, Angew. Chem., Int. Ed., 1998, 37, 2349–2354 CrossRef CAS PubMed
-
CRC Handbook of Optical Resolution via Diastereomeric Salts Formation, ed. D. Kozma, CRC Press, Boca Raton, 2001, ch. 4, pp. 82–87 Search PubMed
-
APEX 2, Version 1.0-27, Bruker AXS Inc., Madison, WI, 2005 Search PubMed
-
SAINT-Plus, Version 7.12, Bruker AXS Inc., Madison, Wisconsin, USA, 2004 Search PubMed
-
G. M. Sheldrick, SADABS: Program for Area Detector Adsorption Correction, University of Göttingen, Germany, 1997, pp. 33–38 Search PubMed
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
- M. Lusi and L. J. Barbour, Cryst. Growth Des., 2011, 11, 5515–5521 CrossRef CAS
- J. S. Bouanga Boudiombo, H. Su, S. A. Bourne, E. Weber and L. R. Nassimbeni, Cryst. Growth Des., 2018, 18, 2670–2677 Search PubMed
- M. Samipillai, E. Batisai, L. R. Nassimbeni and E. Weber, CrystEngComm, 2015, 17, 8332–8338 RSC
- J. S. Bouanga Boudiombo, H. Su, N. Ravenscroft, S. A. Bourne, E. Weber and L. R. Nassimbeni, J. Mol. Struct., 2019, 1181, 636–644 CrossRef CAS
- M. C. Etter, J. C. MacDonald and J. Bernstein, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256–262 CrossRef PubMed
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef CAS
- I. Goldberg, Z. Stein, K. Tanaka and F. Toda, J. Inclusion Phenom. Macrocyclic Chem., 1991, 10, 97–107 CrossRef CAS
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed
- N. B. Báthori and L. R. Nassimbeni, Cryst. Growth Des., 2012, 12, 2501–2507 CrossRef
- M. R. Caira, A. Horne, L. R. Nassimbeni and F. Toda, J. Mater. Chem., 1997, 7, 2145–2149 RSC
- M. R. Caira, A. Horne, L. R. Nassimbeni and F. Toda, J. Mater. Chem., 1998, 8, 1481–1484 RSC
- M. R. Caira, A. Horne, L. R. Nassimbeni and F. Toda, Supramol. Chem., 1998, 9, 231–237 CrossRef CAS
- M. R. Caira, L. R. Nassimbeni, D. Vujovic and F. Toda, J. Phys. Org. Chem., 2000, 13, 75–79 CrossRef CAS
-
G. A. Jeffrey, in Introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S5, Fig. S1–S37. Crystallographic data for this paper have been deposited with the CCDC, accession numbers 1994170 and 1994172–1994174. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce00510j |
| This journal is © The Royal Society of Chemistry 2020 |