
Data mining the Cambridge Structural Database for hydrate–anhydrate pairs with SMILES strings†
Jen E.
Werner
and
Jennifer A.
Swift
 *
*
Georgetown University, Department of Chemistry, Washington, DC 20057-1227, USA. E-mail: jas2@georgetown.edu
First published on 24th March 2020
Abstract
Many organic molecules can crystallize in either hydrated or anhydrous forms. Predicting the formation of hydrates and their relative stability with respect to water-free alternative phases are significant challenges. Here we use the Cambridge Structural Database (CSD) and data informatics to identify and analyze hydrate–anhydrate structure pairs. A search method was developed based on Simplified Molecular-Input Line-Entry strings (SMILES) matching and implemented through the CSD Python Application Programming Interface. Of the >23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecular hydrates containing no metal ions, ∼1400 were found to have at least one corresponding anhydrous form, yielding just over 2000 unique pairs in the CSD. Hydrates with and without a reported anhydrate showed a similar distribution in their water stoichiometries. Lattice symmetry and packing fraction comparisons are reported for the paired hydrates and anhydrates. Structure pairs with one organic component and multiple organic components showed some subtle differences. The details and limitations of the method are outlined in a way that can encourage and guide other types of CSD searches using SMILES.
000 molecular hydrates containing no metal ions, ∼1400 were found to have at least one corresponding anhydrous form, yielding just over 2000 unique pairs in the CSD. Hydrates with and without a reported anhydrate showed a similar distribution in their water stoichiometries. Lattice symmetry and packing fraction comparisons are reported for the paired hydrates and anhydrates. Structure pairs with one organic component and multiple organic components showed some subtle differences. The details and limitations of the method are outlined in a way that can encourage and guide other types of CSD searches using SMILES.
Introduction
It is widely known that crystallization from solution, the preferred method for obtaining most organic molecular crystals, can yield either solvated or solvent-free phases. The most frequently included solvent molecule in molecular crystals is water, making hydrates the largest subclass of solvates.1 The likelihood that an organic molecule will co-crystallize with water remains difficult to predict but is important to control since each crystal form has its own unique set of physical properties.2–4 A third of all pharmaceuticals are estimated to be capable of crystallizing as hydrates.5–8 This estimate in part no doubt reflects the extensive solid form screening efforts required of the industry as well as the desirability of water as a crystallization solvent.Molecular hydrates are often classified based on the topology and local environment of the water molecules within them – as channel hydrates, isolated hydrates and ion-assisted hydrates.9,10 They can alternatively be classified on the basis of their composition – stoichiometric, non-stoichiometric or variable,11 with the exact composition of the latter dependent on the environmental conditions. Hydrates are less frequently classified on the basis of their stability,12–14 particularly in relation to any corresponding anhydrous forms. Some hydrates are stable over a wide temperature range while others spontaneously dehydrate under ambient conditions, either reversibly or irreversibly. Some anhydrous forms are stable in aqueous solution whereas others will quickly recrystallize to a hydrate. The analysis of hydrate stability on a case by case basis remains important, however, this approach has proven rather limited in its ability to more generally predict hydrate properties and hydrate–anhydrate transformations.
The Cambridge Structural Database (CSD), first conceived of over a half-century ago, now contains >1 million structures and is an unmatched repository for small molecule crystallographic data.15 Several previous studies have made use of large bodies of structural data in an effort to glean potential insight into the formation of hydrated phases. These analyses have typically focused on the water binding geometries and topologies within the structure,16–21 rather than on the direct comparison of hydrated and anhydrous forms. The front-end software package ConQuest22 is designed to search the CSD based on chemical name, formula, journal, author, molecular structure or substructure. This makes it well suited for many types of searches, but presents unique challenges related to searching for hydrated and anhydrous structure pairs. ConQuest can easily generate a list of all structures containing water, but there is no efficient way to then search for the corresponding anhydrous form of each molecule without performing literally thousands of individual searches. The development of in-house software to facilitate this kind of search was reported by researchers at the Cambridge Crystallographic Data Center (CCDC) over a decade ago,23,24 though this software was never integrated into the front-end search program.
In 2015, the CCDC introduced the first version of the CSD Python API (Application Programming Interface).25 CSD Python API enables back-end database searching with tailored scripts written to address specific research questions beyond the capabilities of ConQuest. Here we describe how CSD Python API scripts based on Simplified Molecular Input Line Entry strings (SMILES)26 enabled us to search for molecular hydrates, and to identify those with a corresponding anhydrous crystal form in the CSD. The hydrate–anhydrate structure pairs were then evaluated in terms of their packing fraction and lattice symmetry. Statistics generated from all hydrate–anhydrate pairs were compared against a subset of hydrates (with no reported anhydrous form) and a subset of anhydrous phases (with no reported hydrated form).
Representation of organic structures in CSD Python API
A brief discussion of linear notation languages, with a particular focus on Simplified Molecular Input Line Entry strings (SMILES), is necessary to understand our informatics approach. All practicing scientists rely on being able to search many different databases (e.g. PubChem, SciFinder, ChemSpider). A common feature of all chemistry search engines is the need to have some way to convert molecular structures, i.e. the atoms and their connectivity, into a computer-readable format. This is possible through linear notation languages, which define the connectivity through a string of characters. The development of the linear notation language SMILES in the late 1980s was an important step in the advancement of chemical informatics. Other linear notation languages were subsequently developed, including the International Chemical Identifier (InChI),27 which was first publicly introduced in 2005 and is also now in wide use. The specifics of how molecular structures are coded in SMILES and InChI are a bit different, though both notation languages have open source codes28,29 and are incorporated into the CSD Python API.In this work, SMILES strings were used to represent the molecules within a given crystal structure. The basics of molecular representations with SMILES strings are described below, but we recommend David Weininger's original and accessibly written 1988 paper26 for a more detailed description of SMILES methodology. Reading a SMILES string from left to right is equivalent to traversing the atoms of the parent chain in a line-angle drawing in the order in which they are connected. All non-hydrogen atoms are designated by their atomic symbols, the multiplicity of the bond that connects each pair of atoms is indicated with symbols (though single bonds are usually omitted), and any atoms branching off the parent chain are indicated by parentheses. Reading an unsymmetrical molecule from left to right and right to left gives SMILES strings which are recognized as equivalent (e.g. ethanol is CCO or OCC). For cyclic molecules, one bond in the ring is broken and the disconnected atoms are indicated by a number placed after the atomic symbol (e.g. cyclohexane is C1CCCCC1). Because the particular bond broken in a ring is arbitrary, there can be many equivalent SMILES strings for a given cyclic molecule (e.g. tetrahydrofuran is C1OCCC1 or C1COCC1 or O1CCCC1).
Each crystal structure in the CSD has a corresponding entry SMILES string that is composed of the component SMILES string for each molecule in the lattice. When a crystal structure contains more than one molecule, the entry SMILES string separates each component molecule SMILES string with a period. To illustrate, the entry SMILES strings for three different forms of 5-fluorocytosine are shown in Fig. 1.
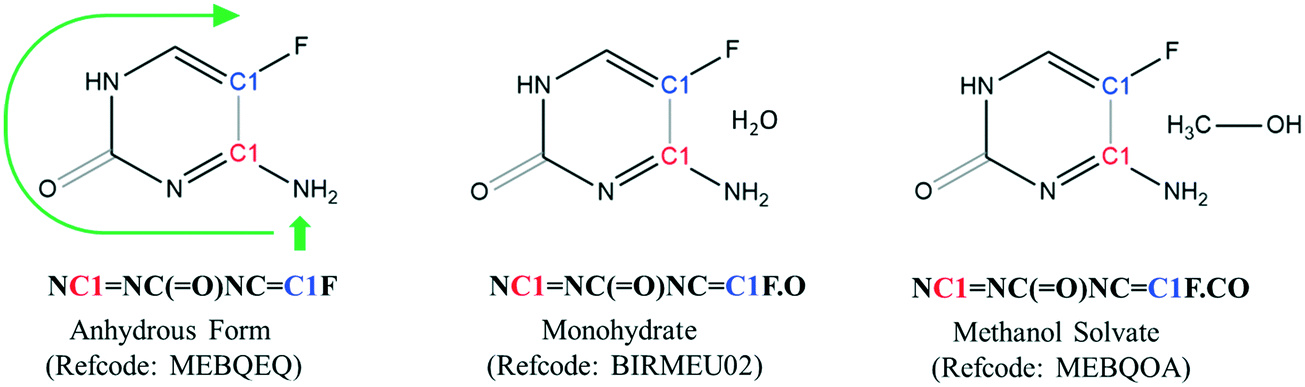 | ||
| Fig. 1 Relationship between molecular structure and the entry SMILES string in three different forms of 5-fluorocytosine. The anhydrous form (refcode: MEBQEQ), monohydrate (BIRMEU02), and methanol solvate (MEBQOA) all contain the same component SMILES string for the organic molecule. | ||
Search strategy overview
Our study employed the Cambridge Structural Database (V5.40, November release) which at the time included just under 1 million crystal structure entries. We restricted our analysis to only organic compounds containing three-dimensional coordinates and no errors. Organometallic compounds and polymers were omitted, as were structures containing any elements aside from H (deuterium isotope allowed), C, N, O, P, S, F, Cl, Br, and I. There were no restrictions on the R-factor and disordered structures were permitted. After application of these criteria, the pool was reduced to a working data set of 327214 structures.
Scripts based on SMILES string matching (and scripts which account for their limitations) were written and implemented in CSD Python API to perform the series of sorting steps shown in Fig. 2. All code used to implement this search is found in ESI.† In step 1, the working data set was first searched for structures which include water in the entry and/or component SMILES string(s). The 24210 structures that had a SMILES string for water were placed in HYDRATE and those that did not were placed in OTHER. However, a small fraction of hydrate structures were not identified in this initial sorting step, either because they have an entry SMILES string of “None” or because the entry SMILES string is incomplete and missing the water component. To capture these missing hydrates, a script was written to perform a text search for structures with the word “hydrate” in the name and H2O1 in the formula within the OTHER list. The text search resulted in 592 additional hydrate entries that were moved to the HYDRATE list. All structures remaining in the OTHER category were renamed ANHYDRATE.
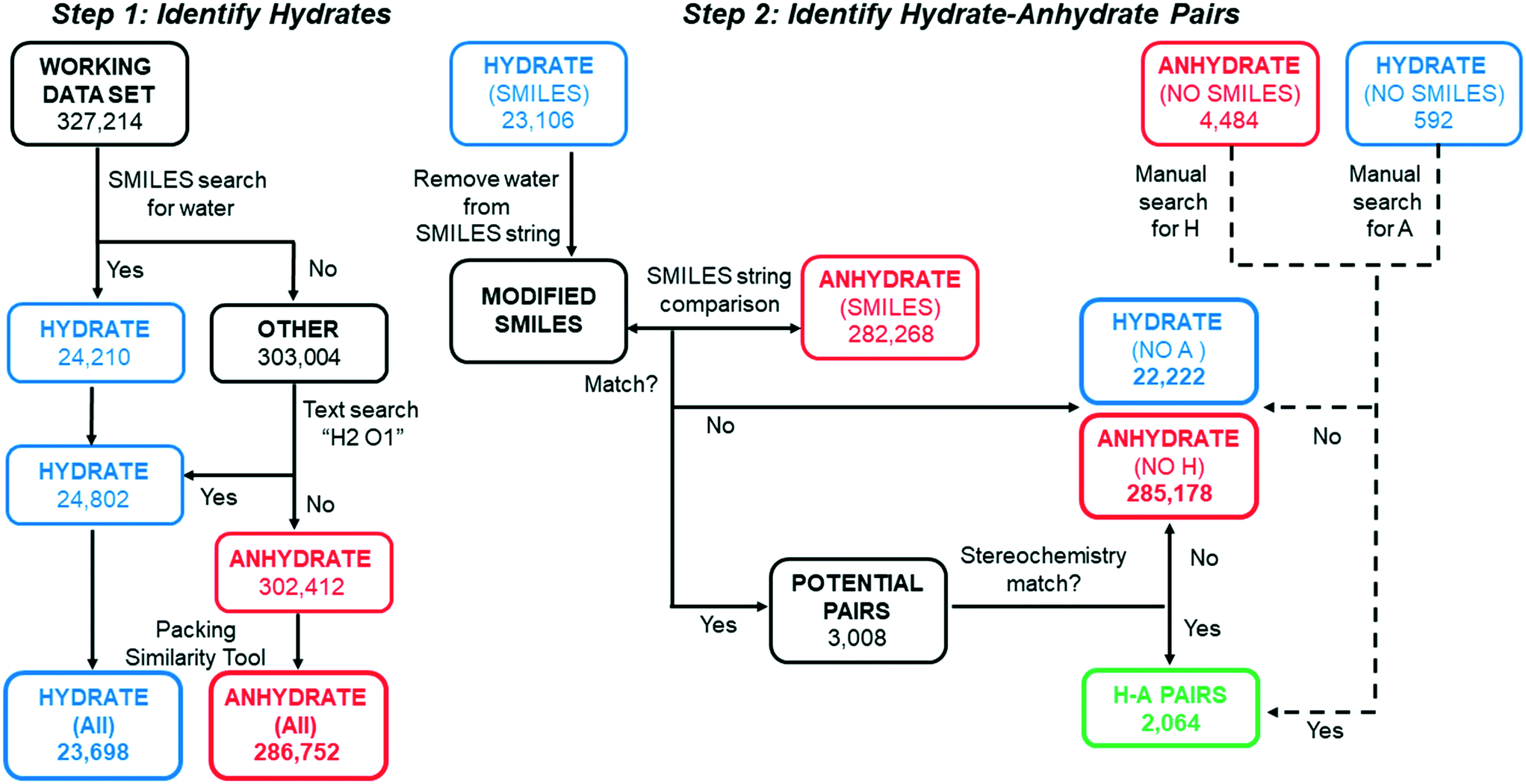 | ||
| Fig. 2 Flow chart illustrating the search steps. The goal of STEP 1 was to identify all molecular hydrate structures in the CSD. The goal of STEP 2 was to identify all unique hydrate–anhydrate pairs. Automated steps are depicted with solid arrows. Manual steps are indicated with dashed arrows. | ||
In cases where two or more refcodes of the same entry existed, the same “Crystal Packing Similarity” function in Mercury was used to determine whether the structures were identical or polymorphs. When an entry in the HYDRATE and ANHYDRATE list was found to be identical, only the first one was retained. In some cases the packing similarity tool could not be used, for example when one of the refcodes was a disordered structure. In those cases, if the entries had unit cell lengths and corresponding angles within 1 Å and 1° of each other, the two refcodes were considered identical. This left 23698 unique structures in the HYDRATE-(All) list and 286752 in the ANHYDRATE-(All) list.
The goal of step 2 was to identify related pairs of hydrates and anhydrates. In order to match structures with the same non-water components, it was important that the SMILES strings generated in Python API account for all components. As previously described, a subset of hydrates structures were found to have incomplete SMILES strings due to a missing water component. Similarly, we found that a subset of other solvates had incomplete entry SMILES strings, due to a missing component string(s) for one or more solvent molecules. To identify and correct solvates with incomplete entry SMILES strings, a text search for the word “solvate” in the name was applied to all structures in the ANHYDRATE-(All) and HYDRATE-(All) lists. The name of each solvent molecule was tagged to its corresponding component SMILES string in a dictionary. If “solvate” appeared in the chemical name, the entry SMILES string was checked to ensure it included all the component SMILES strings corresponding to solvent molecules listed in the name. Any missing solvent molecules were appended from the dictionary to the entry SMILES string, and these corrected SMILES strings were used in subsequent searches.
Two different approaches were needed to find hydrate–anhydrate pairs depending on whether the structure had an entry SMILES string or an entry SMILES string of “None.” Those with entry SMILES strings (97.5% of all hydrates) are considered first. For each structure in the HYDRATE-(SMILES) list, the water component in the string was deleted. Using these MODIFIED SMILES, an automated string-matching search was then performed against the ANHYDRATE-(SMILES) list to identify anhydrous forms with the same organic component(s). (One could alternatively do the reverse – identify anhydrous structures, add water to the SMILES string, then search for hydrates.) If a hydrate–anhydrate match was identified, the structures were put in a list of POTENTIAL PAIRS. If no match was identified, the hydrate and anhydrate structures were placed in HYDRATE-(NO A) and ANHYDRATE-(NO H) individual lists.
The most significant limitation of SMILES string-matching is that it does not account for stereochemistry, so automated searches can over-select for pairs that are not valid. For example, string-matching identifies glucose monohydrate (GLUCMH11) and anhydrous galactose (ADGALA01) as a pair because the sugars have the same component SMILES string (Fig. 3). To identify the false matches in POTENTIAL PAIRS, the configuration of each chiral center in the hydrate and anhydrate molecule(s) was determined with a customized script which mimics the way stereochemistry is distinguished in the InChI notation language. Our script compared the configuration of each tetrahedral atom in the structure as well as the chirality designations (e.g. R, S, rac) in the compound name. Both of these methods require that the chirality is specified in the database entry. If all atoms in the two structures were confirmed to have the same chirality and there was nothing in either chemical name to indicate otherwise, the pair was considered valid and placed in H–A PAIRS. If the components were found to be different stereoisomers, either through their chiral centers or chemical names, the individual entries were redirected to HYDRATE-(NO A) and ANHYDRATE-(NO H).
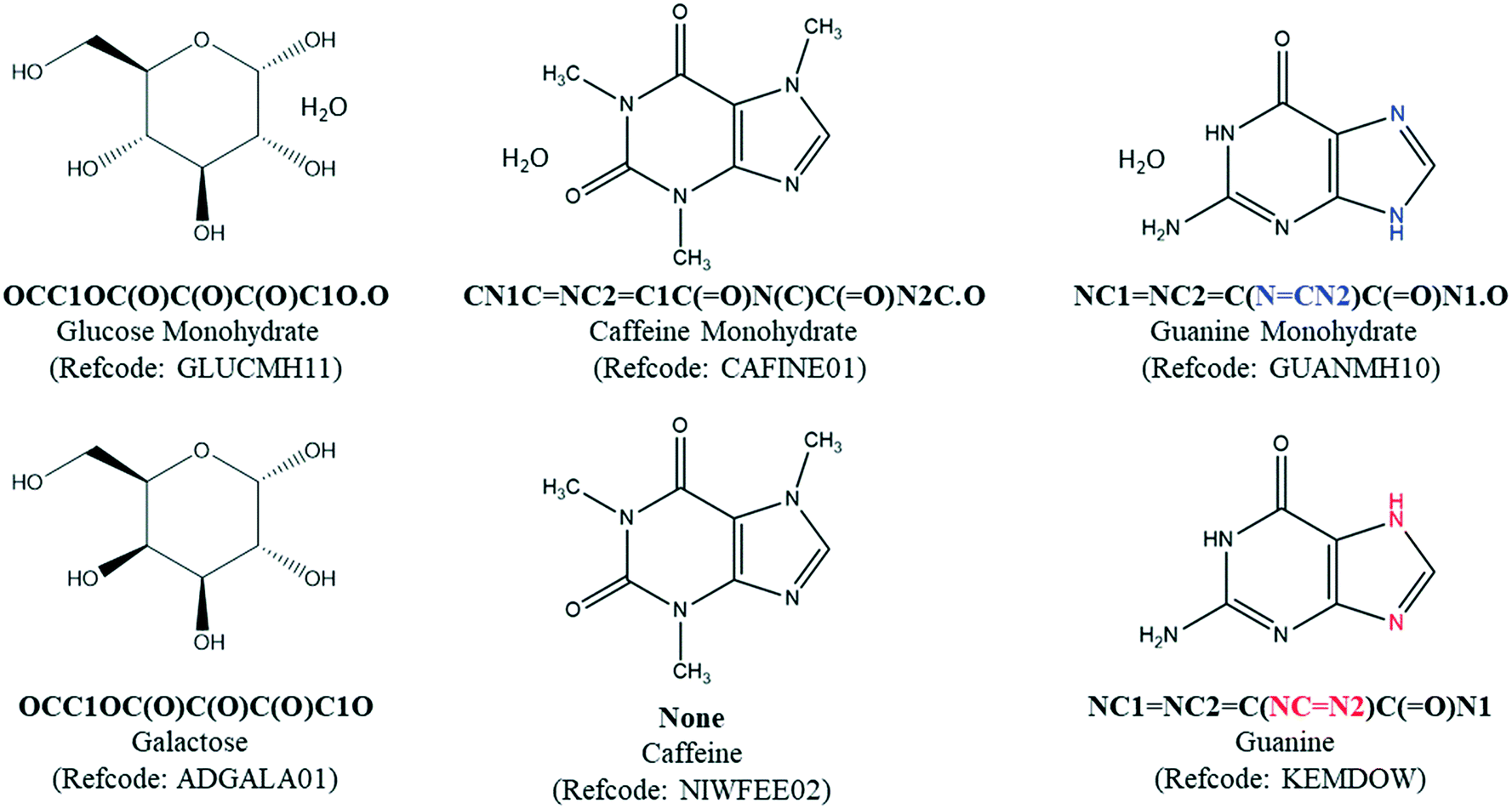 | ||
| Fig. 3 Three general limitations of CSD searches based on SMILES strings include: (1) the absence of stereochemistry, (2) the absence of strings in some CSD entries and (3) different representations for tautomeric molecules. Limitation (1) can lead to over-selection errors, whereas limitations (2) and (3) can lead to under-selection of hydrate–anhydrate pairs. | ||
A different method was needed to search for pairs when the hydrate had an entry SMILES string of “None”. Though a relatively small percentage of structures have this designation, a match cannot be found if either the hydrate or the anhydrate entry SMILES in the pair is “None”. For example, after the water component is removed from the SMILES string of caffeine monohydrate (CAFINE01), the MODIFIED SMILES string would still be unable to match anhydrous caffeine (NIWFEE02) because it has an entry SMILES string of “None”. To correct for this type of under-selection, a Python API script was written to compare the formula of each organic component in the HYDRATE-(NO SMILES) list against entries in the ANHYDRATE-(All) list. Entries with identical chemical formulas were then manually checked and placed in H–A PAIRS if there was a structure match. The same process was carried out for the ANHYDRATE-(NO SMILES) list against the HYDRATE-(All) list. All matches were run through the same chirality script as the H–A PAIRS with entry SMILES strings. These combined searches yielded 2064 H–A PAIRS, which are generated from 1273 unique molecules. The H–A PAIRS list includes cases where a hydrate can correspond to two different anhydrous polymorphs, and each pair is counted. Similarly, an anhydrate can have more than one match if the hydrate is polymorphic or if hydrates with different stoichiometries have been reported.
While the generation of the H–A PAIRS list was done as accurately as possible, there will always be a small number of pairs that are missed or included with an automated search. One class of structure pairs that is potentially under-represented in the search is tautomers. Because tautomers have different SMILES strings, SMILES string matching can not identify these pairs. For example, the double bond and H atom positions in guanine are different in its monohydrate (GUANMH10) and anhydrous (KEMDOW) forms.30 There may be other cases, but we presume the number of other tautomeric hydrate–anhydrate pairs that were not counted in this search is small. In our final manual analysis of the final H–A PAIRS list, we also identified a small number of false hydrate–anhydrate pairs that should have been removed in the stereochemical matching step. Such errors were typically due to missing information in one or both database entries (e.g.inositols MAFSUI and ALITOL and cis/trans isomers EQOWAJ and WOQRIF).
Hydrate stoichiometry
With a list of hydrate–anhydrate pairs generated, the next task was to determine whether there were discernable differences in the hydrates with and without a known anhydrate form. Of the 23698 unique hydrate structures, only 1476 (6.2%) had a corresponding anhydrate in the database, while 22222 (93.8%) did not. Table 1 compares the distribution of water stoichiometries of all hydrates as well as those with and without a corresponding anhydrous form. The stoichiometric sorting was based on the entry formula because SMILES strings cannot distinguish between hydrates with stoichiometric and sub-stoichiometric water content, (e.g. monohydrates and hemihydrates both have one water component).
| Hydrate (all) | Hydrate (NO A) | Hydrate (H–A PAIRS) | |
|---|---|---|---|
| Hydrates with an integral number of water molecules | |||
| Monohydrates | 10977 (46.3%) |
10251 (46.1%) |
726 (49.2%) |
| Dihydrates | 3893 (16.4%) | 3640 (16.4%) | 253 (17.1%) |
| Trihydrates | 1092 (4.6%) | 1037 (4.7%) | 55 (3.7%) |
| Tetrahydrates | 759 (3.2%) | 724 (3.3%) | 35 (2.4%) |
| pentahydrates | 270 (1.1%) | 262 (1.2%) | 8 (0.5%) |
| Hexahydrates | 273 (1.2%) | 260 (1.2%) | 13 (0.9%) |
| Heptahydrates | 113 (0.5%) | 108 (0.5%) | 5 (0.3%) |
| Octahydrates | 145 (0.6%) | 143 (0.6%) | 2 (0.1%) |
| Nonahydrates | 68 (0.3%) | 68 (0.3%) | 0 (0%) |
| Decahydrates | 81 (0.3%) | 79 (0.4%) | 2 (0.1%) |
| More than 10 | 467 (1.9%) | 455 (2.0%) | 12 (0.8%) |
| Hydrates with a non-integral number of water molecules | |||
| Hemihydrates | 2414 (10.2%) | 2247 (10.1%) | 167 (11.3%) |
| Less than 1 | 1242 (5.2%) | 1141 (5.1%) | 101 (6.8%) |
| More than 1 | 1836 (7.7%) | 1740 (7.8%) | 96 (6.5%) |
| Undefined | 68 (0.3%) | 67 (0.3%) | 1 (0.1%) |
| Total |
23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 698 698
|
22222
|
1476 |
Across all hydrate structures, 76.5% were found to have a stoichiometric ratio of water molecules per organic, with monohydrates (46.3%) and dihydrates (16.4%) representing the largest classes. Generally, as the number of water molecules per organic increases, the relative abundance of structures with that stoichiometry decreases. An unexpected trend was observed in the hydrates with >5 water molecules. Structures with an even number of waters (hexa-, octa- and decahydrates) occurred with slightly greater frequency than those with an odd number of waters (penta-, hepta- and nonahydrates). This preference likely stems from water's propensity to form discrete hydrogen bonded clusters and/or extended networks, the majority of which have previously been shown to involve an even number of water molecules in the repeat unit.18,19 The 23.5% of hydrates with a non-integral ratio of water molecules per organic were sorted into four categories: hemihydrates, <1 water per organic, >1 water per organic, and undefined. Hemihydrates (10.2%) were the largest class in this category, and the third largest category overall.
Trends in the hydrates with and without known anhydrous forms are similar, though the data suggests that those with a known anhydrate form are slightly more likely to have 2 or fewer waters per organic molecule. Hydrates with a paired anhydrate in the CSD were ∼ 5% more likely to be hemihydrates, monohydrates, and dihydrates compared to hydrates without an anhydrate form.
Comparing hydrate–anhydrate structure pairs
By identifying systems that are capable of growing in both hydrate and anhydrate forms, it becomes possible to probe whether hydrates and anhydrates exhibit any inherent differences in terms of their lattice symmetry and packing fraction. For some analyses it proved instructive to separate the 2064 unique hydrate–anhydrate pairs into two categories – those with one organic component (1530) and those with more than one organic component (534). In some analyses where the number of pairs meeting certain additional criteria was low, we looked for trends in the unpaired hydrate and anhydrate data sets. Comparison sets of 2000 unpaired hydrates and anhydrates were generated from the 22222 entries in the hydrate-(No A) and the 285178 entries in the anhydrate-(No H) list by selecting alphabetically the first entries with either 1 or 2+ organic components and no disorder.
Lattice symmetry
Each entry in the CSD contains a tag for the lattice symmetry which can be accessed with Python API, which simplified sorting the entries. Table 2 shows the distribution across the seven Bravais lattices in the working data set (after the removal of duplicates) as well as different subcategories of paired and unpaired hydrates and anhydrates. Consistent with many previous analyses of space groups, >90% of structures have triclinic, monoclinic and orthorhombic symmetries.31,32 Comparison of hydrates and anhydrates in the H–A PAIRS list showed that the former were ∼5.5% more likely to adopt triclinic symmetry than the latter (21.3% vs. 15.8%). The comparison sets generated from the unpaired structures confirmed a similar bias with hydrates more likely to have triclinic symmetry than anhydrates (25.6% vs. 22.1%).| Crystal system symmetry | |||||||
|---|---|---|---|---|---|---|---|
| Triclinic | Monoclinic | Orthorhombic | Tetragonal | Trigonal | Hexagonal | Cubic | |
| WORKING DATA SET | 22.4% | 53.0% | 21.8% | 1.3% | 1.1% | 0.3% | 0.09% |
| H–A PAIRS | |||||||
| Hydrates | 21.3% | 50.9% | 20.0% | 2.7% | 2.9% | 1.1% | 1.0% |
| 1 organic | 16.3% | 51.8% | 22.2% | 2.9% | 3.9% | 1.5% | 1.4% |
| 2+ organics | 33.3% | 48.9% | 14.8% | 2.3% | 0.7% | 0% | 0% |
| Anhydrate | 15.8% | 56.5% | 22.4% | 2.0% | 1.8% | 1.2% | 0.2% |
| 1 organic | 13.9% | 56.4% | 23.9% | 2.1% | 2.3% | 1.3% | 0.3% |
| 2+ organics | 20.7% | 56.9% | 18.9% | 1.8% | 0.7% | 1.1% | 0% |
| UNPAIRED | |||||||
| Hydrate-(No A) | 25.6% | 49.2% | 20.4% | 2.1% | 1.6% | 0.8% | 0.3% |
| 1 organic | 18.0% | 51.2% | 25.4% | 2.4% | 1.8% | 1.0% | 0.2% |
| 2+ organics | 31.8% | 47.6% | 16.5% | 1.8% | 1.5% | 0.6% | 0.3% |
| Anhydrate-(No H) | 22.1% | 53.3% | 21.9% | 1.3% | 1.0% | 0.3% | 0.07% |
| 1 organic | 19.8% | 54.4% | 23.6% | 1.2% | 0.8% | 0.3% | 0.03% |
| 2+ organics | 30.2% | 49.7% | 16.2% | 1.6% | 1.7% | 0.4% | 0.2% |
We sought to determine if this bias was unique to water or more generally related to the number of molecules in the unit cell. Hydrates in the H–A PAIRS list inherently have a larger number of molecular components than the corresponding anhydrates. The structures in the H–A PAIRS list were first divided into two categories – those with 1 organic component (1530 pairs) and those with 2+ organic components (534 pairs). Comparisons made between structures in these subcategories yielded even more dramatic differences, with 2+ organic component hydrates twice as likely to be triclinic compared to those with 1 organic component (33.3% vs. 16.3%). The anhydrates in the H–A PAIRS list followed a similar trend where structures with 2+ organic components were about 1.5 times as likely to adopt a triclinic cell compared to those with 1 organic component (20.7% vs. 13.9%).
Hydrates were also sorted according to their water stoichiometry, since increasing water content also increases the number of molecular components in the lattice. Of the 1530 pairs with one organic component, there were reasonable numbers of mono-, di- and trihydrates to draw comparisons. In these cases, the distribution across the various lattice symmetries was the same for each hydrate stoichiometry. Therefore, the decrease in lattice symmetry appears not to correspond to an increase in the total number of components, but rather to an increase in the number of chemically different components. A more detailed analysis of the symmetry elements might help to better elucidate these differences but was beyond the scope of this study. A better understanding of symmetry considerations may have broader implications for the design of functional multicomponent crystals.33,34
To confirm that general trends extracted from this analysis also reflect what occurs on a case-by-case basis, each of the 2064 H–A PAIRS was sorted according to which form had the higher lattice symmetry (Table 3). The same symmetry, most often monoclinic, was found in 960 pairs. The anhydrate had the higher symmetry in 594 pairs, and the hydrate had higher symmetry in 510 pairs. When the crystal symmetry was different – regardless of whether it was the hydrate or anhydrate, it was usually a comparison between (a) triclinic < monoclinic, (b) triclinic < orthorhombic or (c) monoclinic < orthorhombic. Interestingly, the relative occurrence of these scenarios differed in 1 and 2+ organic component systems. In the systems with 1 organic component the higher symmetry structure was most often orthorhombic and the lower monoclinic (case c). In the 2+ component systems the higher symmetry structure was most often monoclinic (case a). Although there are slight differences in the distribution of hydrates and anhydrates across the Bravais lattices in the 1 and 2+ component crystal lists (Table 2), this does not account for the trends observed.
| Same crystal symmetry | No. pairs | |||
|---|---|---|---|---|
| Triclinic | Monoclinic | Orthorhombic | ||
| 1 organic | 53 (7.6%) | 494 (70.8%) | 118 (16.9%) | 698 |
| 2+ organic | 56 (21.4%) | 165 (63.0%) | 37 (14.1%) | 262 |
| Anhydrate = higher crystal symmetry | ||||
|---|---|---|---|---|
| Triclinic | Triclinic | Monoclinic | ||
| Monoclinic | Orthorhombic | Orthorhombic | ||
| 1 organic | 143 (33.8%) | 30 (7.1%) | 197 (46.6%) | 423 |
| 2+ organic | 86 (50.3%) | 20 (11.7%) | 45 (26.3%) | 171 |
| Hydrate = higher crystal symmetry | ||||
|---|---|---|---|---|
| Triclinic | Triclinic | Monoclinic | ||
| Monoclinic | Orthorhombic | Orthorhombic | ||
| 1 organic | 115 (28.1%) | 22 (5.4%) | 177 (43.3%) | 409 |
| 2+ organic | 44 (43.6%) | 7 (6.9%) | 39 (38.6%) | 101 |
Packing fraction
Hydrates and anhydrates were also compared in terms of their packing fraction. Structures which exhibit disorder were first removed from the H–A PAIRS list, since this can introduce errors in the calculated packing fraction. This reduced the number of hydrate–anhydrate pairs to 1483. The H–atom positions in all structures were normalized, then the packing fraction of each was calculated using the packing coefficient algorithm in Mercury. Most structures had packing fractions in the 60–80% range as expected.35 We limited our analysis to pairs in which both structures had been determined from data collected at the same temperature in order to avoid differences due to thermal expansion effects. This significantly reduced the available data to 435 hydrate–anhydrate pairs (note: an additional 211 additional structure pairs have a reported temperature of “None,” but these are not considered here).For most structure pairs (397 of the 435), the difference in packing fraction was 5.0% or less. Of these structure pairs, the hydrate had the higher packing fraction in 213 (54%) and the anhydrate had the higher packing fraction in 184 (46%). Fig. 4 plots the number of times the hydrate/anhydrate has the higher packing fraction as a function of the difference between the two structures in each pair. Blue bars correspond to pairs where the hydrate has a higher packing fraction and red bars correspond to pairs where the anhydrate has a higher packing fraction. When binned in increments of 0.5%, the bias appears to be largely independent of the magnitude of the packing fraction difference. This suggests that close packing considerations may inherently provide a small bias favoring hydrate formation.
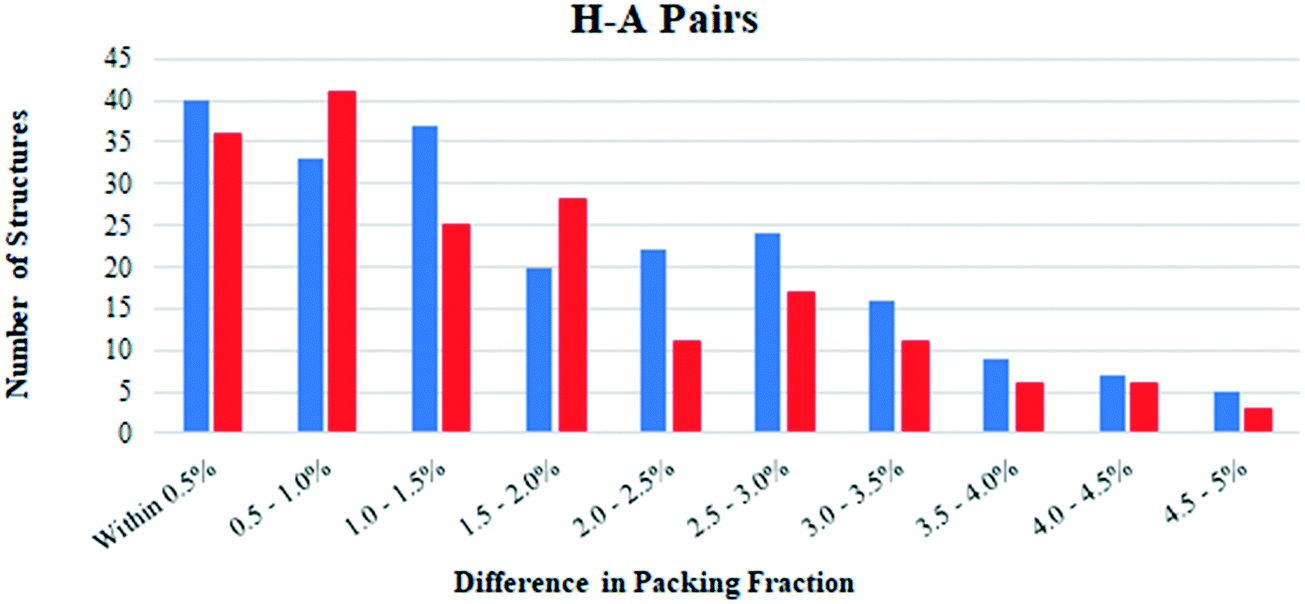 | ||
| Fig. 4 Comparison of the packing fraction differences in hydrate and anhydrate structures in the 417 H–A PAIRS within 5% of each other that were determined at the same temperature. Data is binned in increments of 0.5%. Pairs in which the hydrate and anhydrate have the higher packing fraction are indicated with blue and red bars, respectively. | ||
Conclusions
As the world's largest repository for structural information, there are many new types of questions that can be asked of the CSD. In this report, we have attempted to describe the benefits and some of the limitations of SMILES string matching as a means to identify related structures which are otherwise difficult to identify the existing Conquest software. The hydrate–anhydrate pairs search was accomplished in a two-step process which required many validity checks and the addition of a few unanticipated correction steps.The hydrates with and without anhydrous forms showed nearly identical trends in their water stoichiometries. Compositions with low numbers of water molecules per organic are generally favored (e.g. hemi-, mono-, dihydrates), but in higher hydrates there was a slight preference for compositions with an even rather than odd number of water molecules. In the hydrate–anhydrate pairs, analysis of the distribution across different lattice symmetries and packing fractions also revealed some subtle trends. In hydrate–anhydrate pairs, a bias for hydrates to crystallize in lattices with lower symmetry relative to their anhydrous forms was apparent. Notably, the magnitude of the bias increased with the number of unique molecular components, an observation which has implications beyond the study of hydrates.
The general approach outlined here should be transferrable to other types of searches where the underlying goal is to identify subtle trends across numerous structures. We hope that others with interest in this area can make use of the code provided.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from the National Science Foundation (DMR-1609541) and for a predoctoral fellowship awarded by the Henry Luce Foundation (JEW). We additionally thank Paul Sanschagrin and Stewart Adcock for helpful discussions.Notes and references
- C. H. Görbitz and H.-P. Hersleth, On the inclusion of solvent molecules in the crystal structures of organic compounds, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 526–534 CrossRef PubMed
.
- L. Infantes, L. Fabian and W. D. S. Motherwell, Organic crystal hydrates: what are the important factors for formation, CrystEngComm, 2007, 9, 65–71 RSC
- H. G. Brittain, Polymorphism and Solvatomorphism 2010, J. Pharm. Sci., 2012, 101, 464–484 CrossRef CAS PubMed
- H. D. Clarke, K. K. Arora, H. Bass, P. Kavuru, T. T. Ong, T. Pujari, L. Wojtas and M. J. Zaworotko, Structure−Stability Relationships in Cocrystal Hydrates: Does the Promiscuity of Water Make Crystalline Hydrates the Nemesis of Crystal Engineering?, Cryst. Growth Des., 2010, 10, 2152–2167 CrossRef CAS
- G. P. Stahly, Diversity in Single- and Multiple-Component Crystals. The Search for and Prevalence of Polymorphs and Cocrystals, Cryst. Growth Des., 2007, 7, 1007–1026 CrossRef CAS
- R. K. Khankari and D. J. W. Grant, Pharmaceutical hydrates, Thermochim. Acta, 1995, 248, 61–79 CrossRef CAS
- F. Tian, H. Qu, A. Zimmermann, T. Munk, A. C. Jørgensen and J. Rantanen, Factors affecting crystallization of hydrates, J. Pharm. Pharmacol., 2010, 62, 1534–1546 CrossRef CAS PubMed
- A. M. Healy, Z. A. Worku, D. Kumar and A. M. Madi, Pharmaceutical solvates, hydrates and amorphous forms: A special emphasis on cocrystals, Adv. Drug Delivery Rev., 2017, 117, 25–46 CrossRef CAS PubMed
-
K. R. Morris, Structural aspects of hydrates and solvates, in Polymorphism in Pharmaceutical Solids, ed. H. G. Brittain, Marcel Dekker, Inc., New York, 1999, pp. 125–181 Search PubMed
-
K. R. Morris and N. Rodriguez-Hornedo, in Encyclopedia of Pharmaceutical Technology, ed. J. Swarbrick and J. C. Boylan, Dekker, New York, 1993, vol. 7, pp. 393–441 Search PubMed
-
U. J. Griesser, The importance of solvates, in Polymorphism in the Pharmaceutical Industry, ed. R. Hilfiker, Wiley-VCH, Weinheim, 2006, pp. 211–257 Search PubMed
- S. Gal, Die Wasserdampf-Sorptionsisothermen fester Sorbentien, Chimia, 1968, 22, 409–448 CAS
- S. Petit and G. Coquerel, Mechanism of Several Solid-Solid Transformations between Dihydrated and Anhydrous Copper(II) 8-Hydroxyquinolinates. Proposition for a Unified Model for the Dehydration of Molecular Crystals, Chem. Mater., 1996, 8, 2247–2258 CrossRef CAS
- A. K. Galwey, Structure and order in thermal dehydrations of crystalline solids, Thermochim. Acta, 2000, 355, 181–238 CrossRef CAS
- R. Taylor and P. A. Wood, A Million Crystal Structures: The Whole Is Greater than the Sum of Its Parts, Chem. Rev., 2019, 119(16), 9427–9477 CrossRef CAS PubMed
- G. A. Jeffrey, Water structure in organic hydrates, Acc. Chem. Res., 1969, 2, 344–352 CrossRef CAS
- A. L. Gillon, N. Feeder, R. J. Davey and R. Storey, Hydration in molecular crystals - A Cambridge Structural Database Analysis, Cryst. Growth Des., 2003, 3, 663–673 CrossRef CAS
- L. Infantes and S. Motherwell, Water clusters in organic molecular crystals, CrystEngComm, 2002, 4, 454–461 RSC
- L. Infantes, J. Chisholm and S. Motherwell, Extended motifs from water and chemical functional groups in organic molecular crystals, CrystEngComm, 2003, 5, 480–486 RSC
- A. M. Banaru and Y. L. Slovokhotov, Crystal hydrates of organic compounds, J. Struct. Chem., 2015, 56, 967–982 CrossRef CAS
- R. E. Skyner, J. B. O. Mitchell and C. R. Groom, Probing the average distribution of water in organic hydrate crystal structures with radial distribution functions (RDFs), CrystEngComm, 2017, 19, 641–652 RSC
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, New software for searching the Cambridge Structural Database and visualizing crystal structures, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef PubMed
- J. van de Streek and S. Motherwell, New software for searching the Cambridge Structural Database for solvated and unsolvated crystal structures applied to hydrates, CrystEngComm, 2007, 9, 55–64 RSC
- J. van de Streek, All series of multiple solvates (including hydrates) from the Cambridge Structural Database, CrystEngComm, 2007, 9, 350–352 RSC
- P. Sanschagrin, Using the CSD Python API for interactive analytics and data mining of the Cambridge Structural Database, Acta Crystallogr., Sect. A: Found. Adv., 2017, 73, a67 Search PubMed
- D. Weininger, SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS
- S. R. Heller, A. McNaught, I. Pletnev, S. Stein and D. Tchekhovskoi, InChI, the IUPAC International Chemical Identifier, J. Cheminf., 2015, 7, 23–23 Search PubMed
- http://opensmiles.org/, (accessed July 10, 2019).
- https://www.inchi-trust.org/downloads/, (accessed July 10, 2019).
- A. Hirsch, D. Gur, I. Polishchuk, D. Levy, B. Pokroy, A. J. Cruz-Cabeza, L. Addadi, L. Kronik and L. Leiserowitz, “Guanigma”: The Revised Structure of Biogenic Anhydrous Guanine, Chem. Mater., 2015, 27, 8289–8297 CrossRef CAS
- A. D. Mighell, V. L. Himes and J. R. Rodgers, Space-group frequencies for organic compounds, Acta Crystallogr., Sect. A: Found. Crystallogr., 1983, 39, 737–740 CrossRef
- C. P. Brock and J. D. Dunitz, Towards a Grammar of Crystal Packing, Chem. Mater., 1994, 6, 1118–1127 CrossRef CAS
- C. A. Gunawardana and C. B. Aakeröy, Co-crystal synthesis: fact, fancy, and great expectations, Chem. Commun., 2018, 54, 14047–14060 RSC
- D. J. Berry and J. W. Steed, Pharmaceutical cocrystals, salts and multicomponent systems; intermolecular interactions and property based design, Adv. Drug Delivery Rev., 2017, 117, 3–24 CrossRef CAS PubMed
-
A. I. Kitaigorodsky, Molecular Crystals and Molecules, Academic Press, New York, 1973, vol. 29 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ce00273a |
| This journal is © The Royal Society of Chemistry 2020 |