DOI:
10.1039/C8CE01868E
(Paper)
CrystEngComm, 2019,
21, 494-501
Metal–organic frameworks based on tetraphenylpyrazine-derived tetracarboxylic acid for electrocatalytic hydrogen evolution reaction and NAC sensing†
Received
1st November 2018
, Accepted 20th November 2018
First published on 21st November 2018
Abstract
A series of metal–organic frameworks (MOFs), namely {[Cd(H2TCPP)]·2H2O}n (1), {[Pb2(TCPP)]}n (2), {[La(H2TCPP)]·3.7H2O}n (3), {[Sr3(HTCPP)2]·6H2O}n (4), {[Ce(HTCPP)]·H2O}n (5) and {[Mn2(TCPP)]·2.13H2O}n (6), are prepared from 2,3,5,6-(4-carboxyl-tetraphenyl)pyrazine (H4TCPP) with various metal salts under hydrothermal conditions and are well characterized. The structural analysis shows that compound 1 presents a 2-fold interpenetrating structure, which exhibits a uninodal 4-c net with a {66}{6·6·6·6(2)·6(2)·6(2)} topology. Compound 2 shows a 3-nodal (5,6,11)-connected 3D framework with a point symbol of {410}{414·6}{434·621}. Compound 3 features a (4,8)-connected 3D network built from a binuclear La cluster with a point symbol of {416·612}{44·62}2. Compound 4 demonstrates a 5-nodal (4,5,6,7,8)-connected network with a {410·65}{412·69}{418·610}{46}{47·63} topology. Compound 5 exhibits a 3D interlocked eight-connected 20-c net with a point symbol of {378·498·514}, while compound 6 forms a 3,4,4,5,17-connected net with a point symbol of {4·52}2{432·56·684·74·810}{45·5}{46}{48·52}. In addition, almost all the MOFs display significant fluorescence quenching behaviors by the addition of various nitro-aromatic compounds (NACs), especially TNP. More importantly, six new MOFs have been further investigated to evaluate their electrocatalytic activities for the hydrogen evolution reaction (HER). The results demonstrate that all the MOFs exhibit improved performance compared to ligand H4TCPP for the HER with the lowest onset overpotential of 244 mV and the smallest Tafel slope of 248 mV dec−1, respectively.
Introduction
Metal–organic frameworks (MOFs) are a new class of crystalline porous materials constructed from metal ions or metal ion clusters and bridging organic linkers via hydrothermal synthesis, which have attracted tremendous attention over the last two decades due to their advantages of tunable composition, easy functionalization, accessible metal sites, high drug/gas loading capacity, and good biocompatibility and biodegradability.1 To date, much effort has been made to explore the applications of MOFs, which were extended to a broad range of fields, such as gas storage and separation, catalysis, drug delivery, sensors, etc.2
Due to the serious environmental pollution and the crisis of energy depletion, scientists made great efforts to explore clean and sustainable energy carriers as substitutes for fossil fuels.3 And, hydrogen is considered as one of the ideal alternatives due to its high energy density, zero-emission and non-toxicity.4 As such, electrocatalysis of the hydrogen evolution reaction (HER) via water splitting emerged as a promising approach for chemically offering carbon-free energy and gradually attracted worldwide attention.5 Since the electrocatalytic HER was firstly reported by Kita in 1966, extensive research efforts have been focused on this topic.6 Meanwhile, these research findings indicate that the performance of electrocatalysis is generally limited by the active center, conductivity and pore structure of used materials.7 Up to now, the development of a rapid and efficient method to obtain hydrogen is still highly needed but challenging. One of the promising applications of MOF materials is to use them as electrocatalysts for the HER. In particular, the unique structure of MOFs made them extremely promising precursors/templates for the electrocatalytic HER and great achievements have been made during the last decade. For example, some MOF-derived materials exhibited enhanced electrocatalytic performances, which were superior to noble-metal-based electrocatalysts.8 However, the development of MOF-derived electrocatalysts for the HER is still in its primary stage. Moreover, developing neat MOF-based active materials without conductive additives or other binders for the HER offers challenging issues, which may be ascribed to their poor electrical conductivity.9 On the other hand, in consideration of the human survival environment and health security, exploitation of functional chemosensors for detecting nitro-aromatic compounds (NACs) that can harm individuals and the environment is of great significance, and much effort has recently been devoted to the development of MOF-based chemosensors for the detection of NACs.10
In recent years, we have designed and prepared several porous MOFs that presented fantastic topologies and promising optical properties.11 In our continuing research on the preparation and application of MOFs, we herein developed a series of MOFs based on a tetraphenylpyrazine-derived tetracarboxylic acid skeleton and evaluated their electrocatalytic properties for the HER and sensing behaviors towards NACs. The results indicated that compounds 2 and 5 exhibited promising electrocatalytic performances for the HER while almost all compounds showed obvious fluorescence quenching to NACs, which may provide a new avenue for exploring MOF materials in electrocatalytic and sensing materials.
Experimental
Materials and methods
The tetracarboxyl ligand H4TCPP was synthesized using literature methods.12 All other reagents and solvents commercially available were employed and used without further purification. Elemental analyses were achieved with a Perkin-Elmer 240 elemental analyzer. IR absorption spectra were measured using KBr pellets on a Nicolet 6700 in the range of 400–4000 cm−1 (Fig. S1–S6†). Powder X-ray diffraction (PXRD) patterns were obtained using a Dmax/Ultima IV X-ray powder diffractometer and simulated powder X-ray diffraction patterns were obtained based on single-crystal data using the Mercury software package. Thermal analyses were implemented on a Netzsch STA 449 F3 thermal analyzer and the samples were heated at a rate of 20 °C min−1 from room temperature to 600 °C under an Ar atmosphere. Nitrogen adsorption isotherms were measured at 77 K by using an Autosorb-IQ gas analyzer. The specific surface area and pore size distribution were calculated using the Brunauer–Emmett–Teller (BET) method. Luminescence spectra for the solid samples and liquid samples were recorded on a Cary Eclipse fluorescence spectrophotometer at ambient temperature. UV-vis spectra were recorded on a PerkinElmer Lambda 35 UV-vis spectrometer. Electrochemical measurements were carried out on a computer controlled CHI-660E electrochemical workstation using a standard three electrode configuration consisting of a glassy carbon electrode (GCE) with a diameter of 3 mm as the working electrode, platinum wire as the counter electrode and Ag/AgCl as the reference electrode in an electrolyte solution of 1.0 M KOH (pH = 14). All measurements were conducted at room temperature unless otherwise stated.
Synthesis of {[Cd(H2TCPP)]·2H2O}n (1).
A mixture of H4TCPP (0.05 mmol, 28.1 mg) and Cd(NO3)2·6H2O (0.10 mmol, 31 mg) were dissolved in 14 mL of CH3CN/H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) in a 20 mL vial and heated in a Teflon-lined steel bomb at 160 °C for 4 days. Yellow block crystals were collected in 61.0% yield (based on H4TCPP). Anal. calcd. for C32H16CdN2O10 (%): C, 54.84; H, 2.30; N, 4.00. Found (%): C, 54.90; H, 2.36; N, 4.12%. IR (KBr, cm−1): 3416.39 (s), 1580.27 (s), 1541.81 (s), 1387.96 (s), 1096 (w), 1110.03 (m), 856.19 (m).
:3) in a 20 mL vial and heated in a Teflon-lined steel bomb at 160 °C for 4 days. Yellow block crystals were collected in 61.0% yield (based on H4TCPP). Anal. calcd. for C32H16CdN2O10 (%): C, 54.84; H, 2.30; N, 4.00. Found (%): C, 54.90; H, 2.36; N, 4.12%. IR (KBr, cm−1): 3416.39 (s), 1580.27 (s), 1541.81 (s), 1387.96 (s), 1096 (w), 1110.03 (m), 856.19 (m).
Synthesis of {[Pb2(TCPP)]}n (2).
A mixture of H4TCPP (0.05 mmol, 28.1 mg) and Pb(NO3)2 (0.10 mmol, 22 mg) were dissolved in 14 mL of CH3CN/H2O (4:3) in a 20 mL vial and heated in a Teflon-lined steel bomb at 160 °C for 4 days. Colorless crystals were collected in 53.0% yield (based on H4TCPP). Anal. calcd. for C32H16N2O8Pb2 (%): C, 39.51; H, 1.86; N, 2.88. Found (%): C, 39.53; H, 1.88; N, 2.92%. IR (KBr, cm−1): 3735.79 (w), 3474.92 (m), 1638.80 (m).
Synthesis of {[La(H2TCPP)]·3.7H2O}n (3).
A mixture of H4TCPP (0.05 mmol, 28.1 mg) and La(NO3)3 (0.10 mmol, 15 mg) were dissolved in 14 mL of CH3CN/H2O (4:3) in a 20 mL vial and heated in a Teflon-lined steel bomb at 160 °C for 4 days. Colorless crystals were collected in 48.0% yield (based on H4TCPP). Anal. calcd. for C32H19LaN2O11.70 (%): C, 50.73; H, 2.53; N, 3.70. Found (%): C, 50.12; H, 2.58; N, 3.72. IR (KBr, cm−1): 3426.42 (s), 2971.57 (w), 1610.37 (m), 1531.77 (m), 1377.93 (s), 787.63 (w).
Synthesis of {[Sr3(HTCPP)2]·6H2O}n (4).
A mixture of H4TCPP (0.05 mmol, 28.1 mg) and SrCl3·6H2O (0.10 mmol, 12 mg) were dissolved in 14 mL of CH3CN/H2O (4:3) in a 20 mL vial and heated in a Teflon-lined steel bomb at 160 °C for 4 days. Colorless crystals were collected in 57.0% yield (based on H4TCPP). Anal. calcd. for C64H45N4O22Sr3(4) (%): C, 51.70; H, 3.19; N, 3.77. Found (%): C, 51.82; H, 3.22; N, 3.78. IR (KBr, cm−1): 3436.46 (s), 1590.30 (m), 1387.96 (m), 1270.90 (s).
Synthesis of {[Ce(HTCPP)]·H2O}n (5).
A mixture of H4TCPP (0.05 mmol, 28.1 mg) and CeCl3·7H2O (0.10 mmol, 37 mg) were dissolved in 14 mL of CH3CN/H2O (4:3) in a 20 mL vial and heated in a Teflon-lined steel bomb at 160 °C for 4 days. Colorless crystals were collected in 57.0% yield (based on H4TCPP). Anal. calcd. for C32H19N2O9Ce (%): C, 51.55; H, 2.30; N, 3.76. Found (%): C, 51.64; H, 2.38; N, 3.78. IR (KBr, cm−1): 3436.46 (s), 1610.37 (m), 1531.77 (m), 1387.96 (s), 787.62 (w), 730.77 (w).
Synthesis of {[Mn2(TCPP)]·2.13H2O}n (6).
A mixture of H4TCPP (0.05 mmol, 28.1 mg) and MnCl2·6H2O (0.10 mmol, 20 mg) were dissolved in 14 mL of CH3CN/H2O (4:3) in a 20 mL vial and heated in a Teflon-lined steel bomb at 160 °C for 4 days. Light yellow crystals were collected in 43.0% yield (based on H4TCPP). Anal. calcd. for C32H19Mn2N2O10.13 (%): C, 57.68; H, 2.42; N, 4.20. Found (%): C, 57.72; H, 2.43; N, 4.24. IR (KBr, cm−1): 3426.42 (s), 1657.19 (m), 1600.33 (s), 1531.77 (s), 1387.96 (s), 787.63 (w), 710.71 (w).
Crystallographic data collection and refinement
The structural data of 1–6 were collected at 293/273 (K) on a Bruker SMART APEX-II CCD detector with graphite monochromatic Mo-Kα radiation (λ = 0.71073 Å). All of the structures were solved by direct methods and refined with full-matrix least-squares procedures based on F2 using SHELXS-97 and SHELXL 97 programs.13 The non-hydrogen atoms were refined with anisotropic displacement parameters except for some free solvent molecules, and all the hydrogen atom positions were generated geometrically at idealized positions and refined by using a riding model. The topological analyses were performed using the TOPOS program.14 Details of the crystal parameters, data collection, and refinements for compounds 1–6 are summarized in Table 1. The selected bond lengths and angles are listed in Table S1 in the ESI.†
Table 1 Crystallographic data of compounds 1–6
| Complex |
1
|
2
|
3
|
4
|
5
|
6
|
|
R
1 = ∑|Fo| − |Fc‖/∑|Fo|.
wR2 = [∑w(|Fo|2 − |Fc|2)2/∑w(Fo2)2]1/2.
|
| Formula |
C32H16CdN2O10 |
C32H16N2O8Pb2 |
C32H19LaN2O11.70 |
C64H45N4O22Sr3 |
C32H19CeN2O9 |
C32H19Mn2N2 O10.13 |
| Mass |
700.87 |
970.85 |
757.6 |
1484.91 |
715.61 |
703.37 |
| Temperature/K |
293 K |
293 K |
293 K |
273 K |
293 K |
293 K |
| Crystal system |
Orthorhombic |
Triclinic |
Triclinic |
Triclinic |
Triclinic |
Orthorhombic |
| Space group |
Fddd
|
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P |
Pnna
|
|
a (Å) |
12.850(4) |
9.9328(7) |
10.14(2) |
11.9979(14) |
10.124(2) |
19.677(7) |
|
b (Å) |
18.906(6) |
11.8804(9) |
11.23(3) |
17.366(2) |
11.220(2) |
12.830(4) |
|
c (Å) |
61.98(3) |
14.8808(10) |
15.24(3) |
17.573(2) |
15.330(3) |
30.250(11) |
|
α (°) |
90 |
84.722(3) |
96.47(4) |
83.274(5) |
96.448(4) |
90 |
|
β (°) |
90 |
72.677(3) |
108.69(3) |
75.950(5) |
108.756(7) |
90 |
|
γ (°) |
90 |
87.758(3) |
98.73(5) |
82.588(4) |
98.885(7) |
90 |
|
V (Å3) |
15058(9) |
1669.1(2) |
1601(6) |
3508.4(8) |
1604.4(6) |
7637(5) |
|
Z
|
16 |
2 |
2 |
2 |
2 |
8 |
|
D
Calcd. (g cm−3) |
1.237 |
1.932 |
1.571 |
1.406 |
1.481 |
1.224 |
|
F (000) |
5600 |
900 |
751 |
1494 |
710 |
2848 |
| GOOF |
1.030 |
1.074 |
1.063 |
1.315 |
1.226 |
1.033 |
|
μ (mm−1) |
0.629 |
10.122 |
1.399 |
2.346 |
1.472 |
0.711 |
|
R
int
|
4.72% |
9.06% |
5.26% |
5.55% |
3.48% |
7.19% |
|
R
1 [I > 2σ(I)]a |
0.0313 |
0.0350 |
0.0259 |
0.0529 |
0.0220 |
0.0861 |
| wR2 (all data)b |
0.0797 |
0.0939 |
0.0645 |
0.1781 |
0.0742 |
0.2666 |
Results and discussion
Crystal structure of {[Cd(H2TCPP)]·2H2O}n (1)
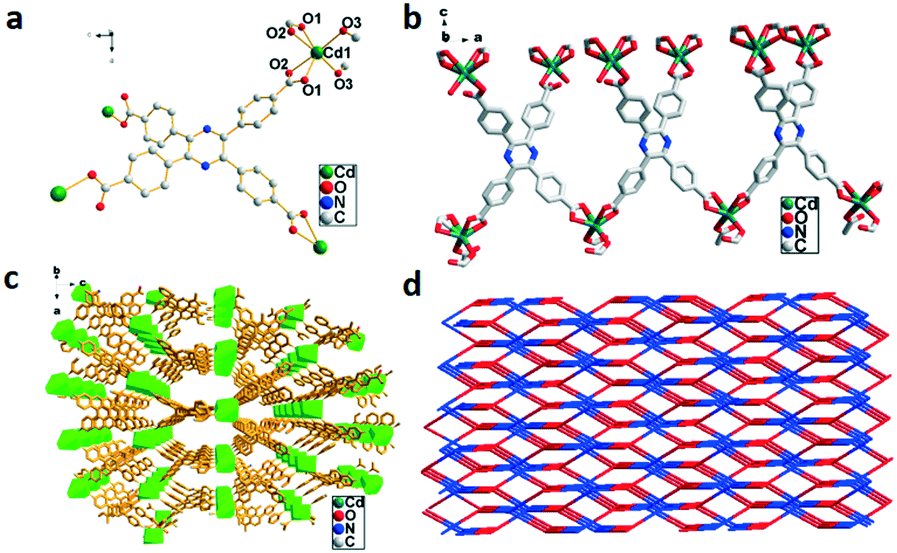
The X-ray structural analysis reveals that 1 belongs to the orthorhombic system with the space group Fddd. The asymmetric unit contains tetranuclear Cd4(TCPP)4 units. Cd2+ centers are six-coordinated by six O atoms from four H4TCPP molecules to form distorted octahedral geometries, as shown in Fig. 1a, with bond lengths in the range of 2.376(4)–2.240(4) similar to those reported in the literature.11c,15 Each ligand coordinates to four Cd2+ centers via two different modes to form a 2D network structure (Fig. 1b). An infinite 3D self-penetrating framework is further formed by the connection of bridge ligands and adjacent Cd2+ centers with a layer distance of 11.4301(26) Å (Fig. 1c). The whole structure displays a 3D 2-fold interpenetrating 6-connected 4-c sqc182 net with a point symbol of {66}{6·6·6·6(2)·6(2)·6(2)} as illustrated in Fig. 1d.
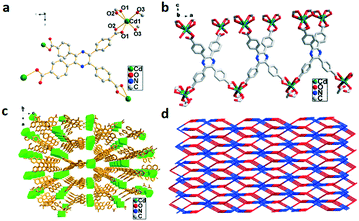 |
| | Fig. 1 (a) Coordination environment of Cd2+ in 1, symmetry code: Cd1 = x, y, z; O2 = 1 − x, 0.25 + y, 0.25 + z. (b) 2D structure of 1 viewed along the b axis. (c) 3D framework of 1. (d) 2-fold interpenetrated net of 1. | |
Crystal structure of {[Pb2(TCPP)]}n (2)
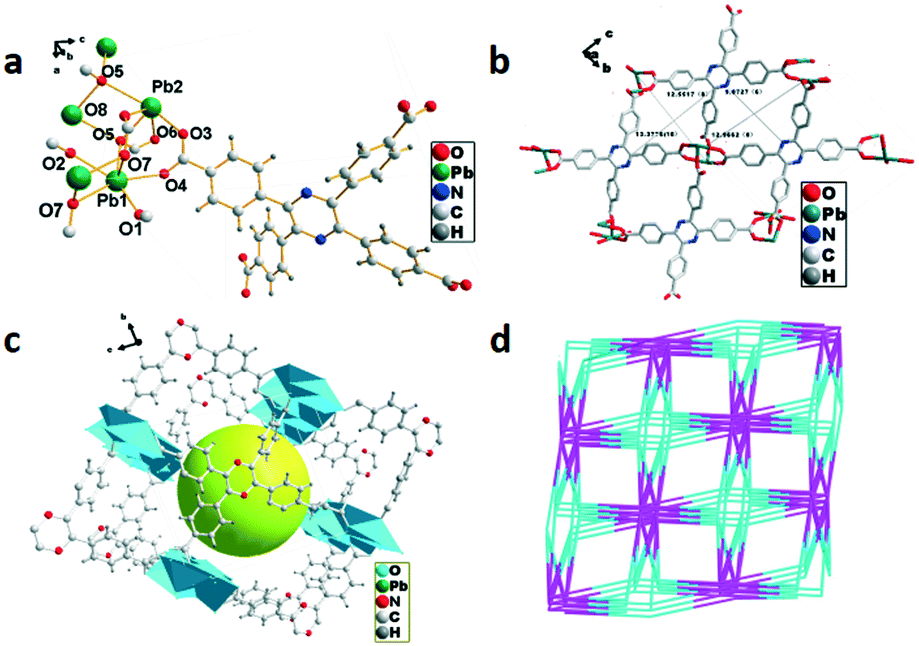
The single-crystal X-ray diffraction analysis reveals that 2 crystallized in the triclinic system with the space group P. As shown in Fig. 2a, two coordination modes of Pb2+ are observed in the asymmetric unit, in which Pb1 adopts a six-coordinated mode with six oxygen atoms from six H4TCPP molecules, while Pb2 displays a five-coordinated mode with five oxygen atoms from four H4TCPP molecules. A parallelogram structure is formed by atoms O7–Pb1–O7–Pb1 in a clockwise direction with an angle ∠O7–Pb1–O7 of 78.828(3)°. Pb1 and Pb2 further connect with H4TCPP to generate a 2D framework along the a axis containing two 1D nanosized quadrilateral channels with dimensions of 11.8775(7) Å × 12.5517(8) Å and 15.3615(10) Å × 11.7945(8) Å, respectively (Fig. 2b). The crystal accumulation diagram exhibits the formation of a hole in the cavity of complex 2 (Fig. 2c). Topologically, the H4TCPP ligand can be regarded as a 5,6-connected node, while Pb2+ belongs to a 11-connected node. Therefore, the whole structure can be simplified as a 3-nodal (5,6,11)-connected topology with a Schläfli notation of {410}{414·6}{434·621} as shown in Fig. 2d.
 |
| | Fig. 2 (a) Coordination environment of Pb2+ in 2, symmetry code: Pb1 = x, y, z; O4 = −x, 1 − y, 1 − z. (b) 2D net structure of 2 with two 1D nanosized quadrilateral channels. (c) 3D framework of 2. (d) Topology structure of 2. | |
Crystal structure of {[La(H2TCPP)]·3.7H2O}n (3)
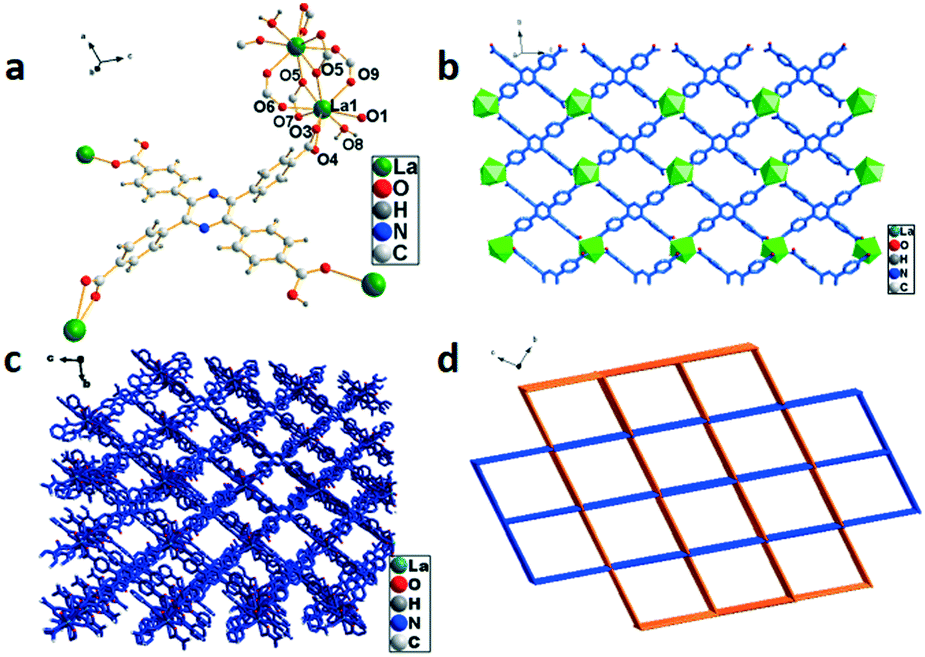
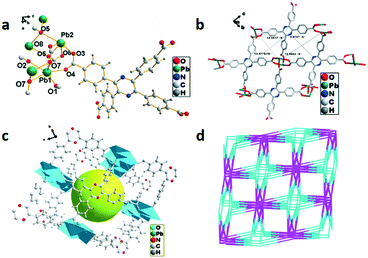
The single-crystal X-ray study reveals that 3 crystallized in the triclinic system with the space group P. A slightly disordered water molecule is observed which causes the non-integer number of oxygen atoms in the molecular formula, and meanwhile the hydrogen atoms cannot be obtained by Fourier differential peak synthesis. The asymmetric unit is composed of four La3+ ions, four H4TCPP molecules and one water molecule as displayed in Fig. 3a, in which La3+ exhibits a distorted decahedral coordination configuration connected by eight oxygen atoms from H4TCPP (La–O, 2.499(4)–2.673(5) Å) and one oxygen atom from water molecules (La–O, 2.531(4) Å). The adjacent La3+ ions bonded to two H4TCPP ligands form a dinuclear {La2} unit and two types of coordination modes can be observed: one employs a bidentate bridging coordination fashion μ2, while the other adopts a monodentate coordination mode via only an oxygen atom of the carboxylic group to a La3+ ion. Further connection of dinuclear {La2} units with H4TCPP ligands generates a 2D structure (Fig. 3b), which self-assembles into 3D networks (Fig. 3c). Topologically, 3 exhibits a 3D (4,8)-connecting 2-nodal 4,8-c net with stoichiometry (4-c)2(8-c) evaluated as an sqc170 topology type, in which La3+ is regarded as an 8-connector. Each {La2} unit is bonded to six H4TCPP ligands, further connecting eight {La2} units, presenting an eight-connected pcu network with a point symbol of {416·612}{44·62}2 (Fig. 3d).
 |
| | Fig. 3 (a) Coordination environment of La3+ in 3, symmetry code: La1 = x, y, z; O5 = x, y, z. (b) 2D structure of 3. (c) Crystal accumulation diagram of 3. (d) Topology structure of 3. | |
Crystal structure of {[Sr3(HTCPP)2]·6H2O}n (4)
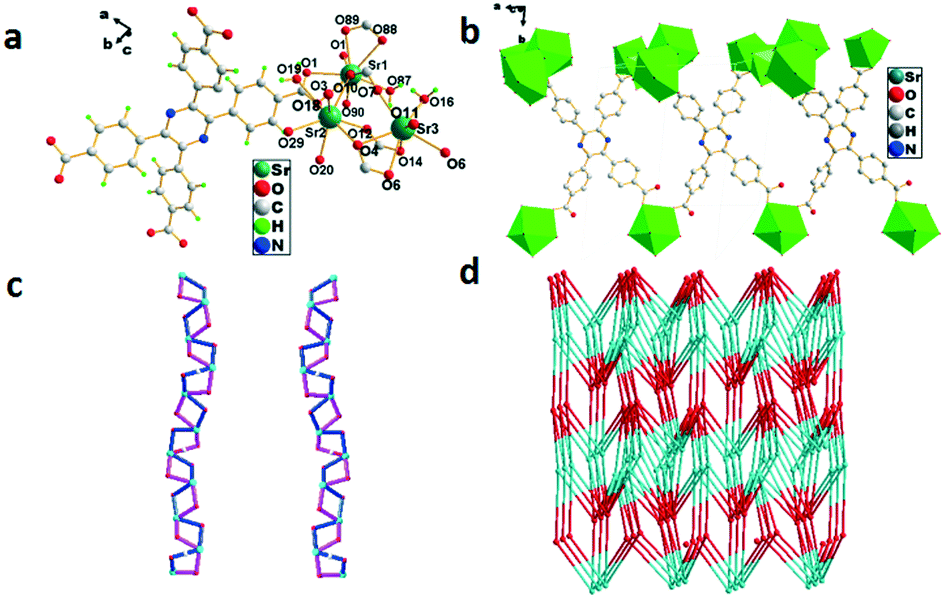
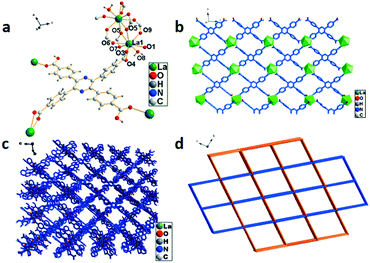
The single-crystal X-ray analysis shows that 4 crystallized in the triclinic system with the space group P. Sr1 is linearly eight-coordinated by six oxygen atoms from four H4TCPP molecules and two oxygen atoms from solvent water molecules with bond lengths of 2.508(2)–2.717(2) Å (Sr1–O) (Fig. 4a). Both Sr2 and Sr3 adopt a distorted octahedral geometry coordinated by four oxygen atoms from four H4TCPP molecules and two oxygen atoms from solvent water molecules with bond lengths in the range of 2.518(2)–3.005(3)Å (Sr–O) (Fig. 4b). An attractive Sr-cam double helical chains can be observed in the left-handed (blue) and right-handed (violet) helical chains intertwined with each other along the b axis. The distances of two adjacent Sr3+ centers were calculated to be 4.1101(4) Å and 3.9685(4) Å, respectively (Fig. 4c). Topologically, 4 can be rationalized as a 3-D (4,5,6,7,8)-connected network with a point symbol of {410·65} {412·69}{418·610}{46}{47·63} in the case that Sr3+ is regarded as an 8-connector (Fig. 4d).
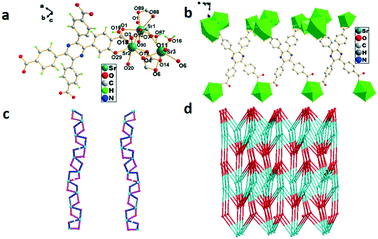 |
| | Fig. 4 (a) Coordination environment of Sr3+ in 4, symmetry code: Sr1 = x, 1 + y, z; Sr2 = 1 − x, 1 − y, 1 − z. (b) Sr3+ ion polyhedral diagram of 4. (c) The left-and right-handed helical chains of Sr-cam. (d) 3D 5-nodal (4,5,6,7,8)-connected topological network. | |
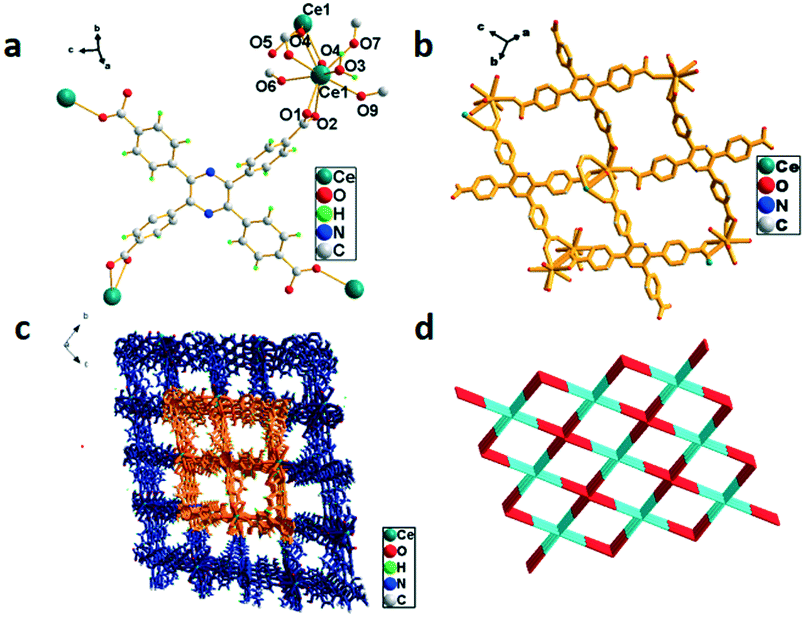
Crystal structure of {[Ce(HTCPP)]·H2O}n (5)
The X-ray structural analysis reveals that 5 crystallized in the triclinic system with the space group P. The asymmetric unit contains two Ce3+ ions, one H4TCPP molecule and a water molecule, as shown in Fig. 5a. Ce3+ ions adopt a nine-coordinated mode to form a distorted decahedron, in which Ce3+ is surrounded by eight oxygen atoms from six H4TCPP molecules and another oxygen atom from a water molecule. A binuclear cluster unit (Ce1–O4–Ce1–O4) forms by the connection of two Ce atoms via two oxygen atom (O4) bridges, and the distance of Ce(1)⋯Ce(1) is 4.1223(9) Å. The H4TCPP connects four Ce(III) ions and the adjacent Ce(III) ions are bridged by two H4TCPP molecules so as to form 2D-cavity with sizes of ca. 10.3113(17) Å × 15.3304(25) Å and 12.9555(20) Å × 12.8338(21) Å along the b axis, respectively (Fig. 5b). The {Ce2} units further connect with another two H4TCPP molecules to form a 3D porous structure as shown in Fig. 5c. Topological analysis suggests that 5 exhibits a uninodal 8-connected 20-c net topology with a point symbol of {378·498·514} (Fig. 5d).
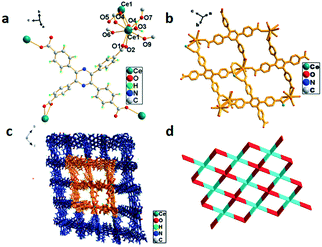 |
| | Fig. 5 (a) Coordination environment of Ce3+ in 5, symmetry code: Ce1 = x, y, z; O1 = x, y, 1 + z. (b) 2D structure of 5. (c) The 3D framework of 5. (d) 3D 8-connected topological network. | |
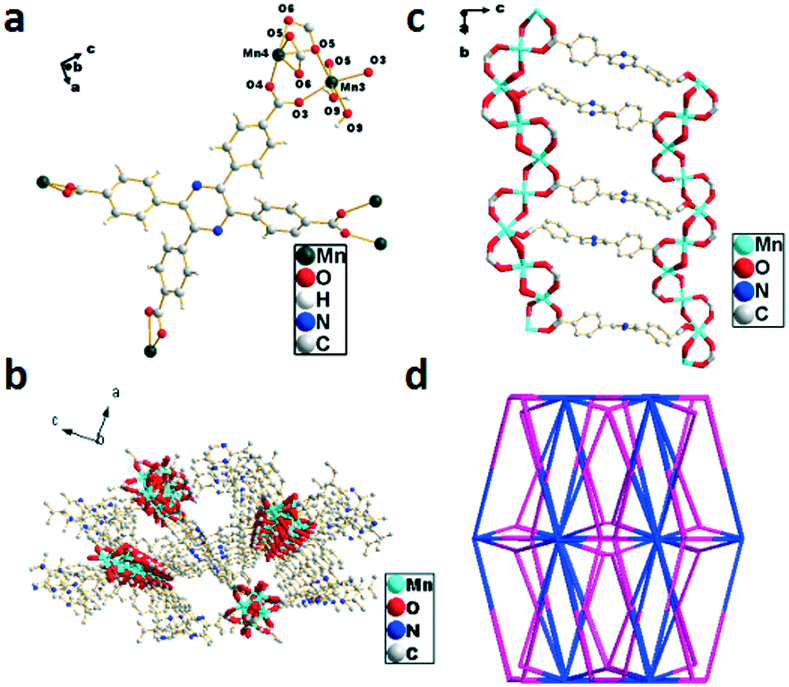
Crystal structure of {[Mn2(TCPP)]·2.13H2O}n (6)
The single-crystal X-ray diffraction analysis reveals that 6 crystallized in the monoclinic system with the space group Pnna. The non-integer number of the oxygen atom in molecule 6 can be ascribed to the same reason as that depicted in 3. The asymmetric unit contains two Mn(II) ions and one H4TCPP molecule to form a distorted octahedral fashion, which consists of six oxygen atoms from four H4TCPP molecules with Mn–O bond lengths in the range of 2.027–2.319 Å (Fig. 6a). The deprotonated carboxylic acid group of H4TCPP coordinates to Mn2+ ions via two different modes resulting in the formation of a ladder-shaped network, in which the two helical chains and ligands were seen as banisters and staircases, respectively (Fig. 6b). Each 2D chain interacts with neighboring chains via the H4TCPP ligand, which further extends the 2D framework to a 3D supramolecular framework (Fig. 6c). Topologically, the whole structure can be simplified as a 3,4,4,5,17-connected net with a point symbol of {4·52}2{432·56·684·74·810}{45·5}{46}{48·52} (Fig. 6d).
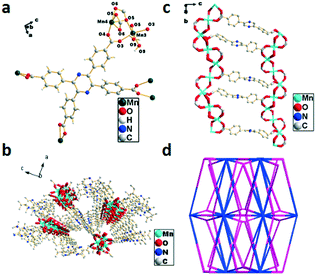 |
| | Fig. 6 (a) Coordination environment of Mn2+ in 6, the hydrogen atoms are omitted for clarity, symmetry code: Mn3 = x, y, z; Mn4 = x, y, z. (b) Double-helix chain of 6. (c) Views of the 3D framework of 6 along the b axis. (d) Topological structure of 6. | |
PXRD, thermogravimetric analysis and adsorption properties
PXRD shows that the peak positions of the obtained crystal samples of 1–6 are in excellent agreement with the simulated peak positions from the single crystal data, which indicates the phase purity of the synthesized samples (Fig. S7–Fig. S12†). TG on 1–6 was investigated in the temperature range of 0–600 °C (Fig. S13†). The thermogravimetric curve displays that 1 decomposes in three obvious weight loss steps, including the initial weight loss of 7.6% from 0–160 °C ascribed to residual water molecules in the crystal, the weight loss of 4.4% (calcd 4.5%) of two coordinated water molecules from 160–440 °C and the weight loss at around 440 °C for structural decomposition of the MOF framework. 2 lost one lattice water molecule from 0 to 100 °C with a weight loss of 1.4% (calcd. 1.6%), and then remained thermally stable till 430 °C without any weight loss. 3 lost one lattice water molecule from 0–102 °C with a weight loss of 2.2% (calcd. 2.3%) and two coordinated water molecules at 102–420 °C with a weight loss of 4.2% (calcd. 4.4%), and then began to collapse at around 420 °C. 4 exhibited a weight loss of 2.6% (calcd. 3.0%) from 0–400 °C corresponding to three coordinated water molecules. 5 lost one coordinated water molecule from 0–200 °C with a weight loss of 2.1% (calcd. 2.2%), and then remained thermally stable till 432 °C. 6 presented a weight loss of 4.8% (calcd. 5.1%) ascribed to two lattice water molecules from 120–430 °C, then the whole structure began to collapse at around 430 °C. The thermogravimetric analyses suggest that all MOFs exhibit excellent thermal stability. Subsequently, the porosity of 1–6 was investigated via N2 adsorption at 77 K. As shown in Fig. S14,†3 and 6 achieve saturated N2 uptake values of 46 and 40 cm3 g−1 (STP), and the calculated Brunauer–Emmett–Teller (BET) surface areas are 18.325 m2 g−1 and 31.128 m2 g−1, respectively. The experimental total pore volumes for 3 and 6 were calculated to be 0.06396 cm3 g−1 and 0.07064 cm3 g−1, respectively. However, the other MOFs showed relatively poor performance in N2 adsorption, which could be mainly ascribed to their BET surface areas and pore volumes, and the corresponding data are listed in Table S2.†
Optical properties and NAC sensing
The UV/vis and emission spectra of all MOFs as well as the ligand H4TCPP were investigated in the solid state at room temperature (Fig. S15 and S16†). As can be seen in the UV-vis absorption spectra, H4TCPP and complexes 1–6 showed similar absorption bands in the UV regions of 220–310 nm and 310–400 nm, which could be ascribed to the π–π* transition between ligands and metal-to-ligand charge-transfer (MLCT) transitions.16 In addition, the solid state fluorescence spectra of 1–6 as well as H4TCPP are shown in Fig. S16.† The results indicated that compound H4TCPP exhibited the maximum emission band centered at 467 nm, while the maximum emissions of compounds 1–4 showed obvious blue shifts compared to that of H4TCPP, which may be assigned to the intraligand π–π* or n–π* transition.11a,17 However, compounds 5 and 6 show no obvious characteristic emission peak, which may be ascribed to several factors, such as different coordination modes, coordination environments of metal ions and rigid solid-state crystal packing.18
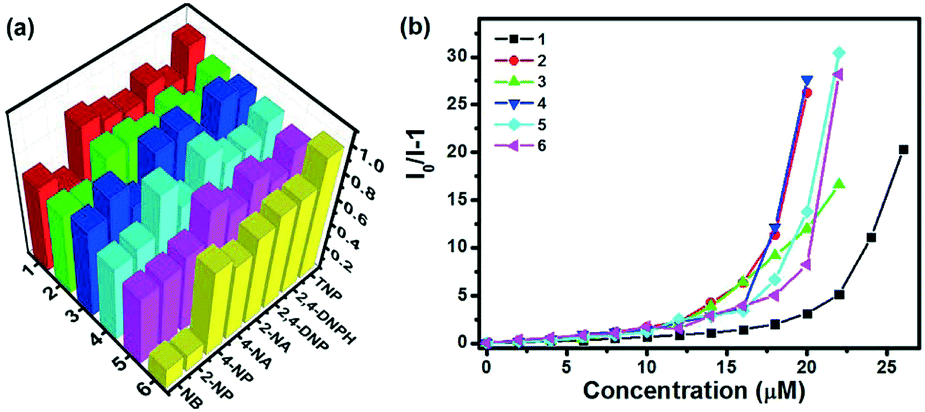
Subsequently, all MOFs were further explored for sensing nitro-aromatic compounds (NACs), a class of highly toxic and explosive materials. The fluorescence sensing performance of MOFs 1–6 towards various NACs was investigated by the addition of identical amounts of NACs, such as 2-nitroaniline (2-NA), 4-nitroaniline (4-NA), 2-nitrophenol (2-NP), 4-nitrophenol (4-NP), nitrobenzene (NB), 2,4-dinitrophenol (2,4-DNP), 2,4-dinitrophenylhydrazine (2,4-DNPH) and picric acid (TNP) to various MOFs dispersed in water. As shown in Fig. 7a, all MOFs showed high selectivity to TNP with ratios of fluorescence quenching in the range of 87–100%, whereas they exhibited relatively poor selectivity to 2-NP and NB with the maximum ratio of fluorescence quenching of 84.8% and 71.8%, respectively. The selectivity may be contributed to the interactions between the open nitrogen atoms in the pyrazine unit and the free acid sites (amino or hydroxyl) of NACs as well as the electron and energy transfer between the electron-deficient TNP and fluorophore.10a,12,19 In order to further compare the efficiency of various sensors, fluorescence titrations were carried out with gradual addition of TNP to various MOFs dispersed in water as depicted in Fig. S17,† and the Stern–Volmer (SV) equation, (I0/I) = Ksv [A] + 1, where I0 is the initial fluorescence intensity before the addition of an analyte, I is the fluorescence intensity in the presence of the analyte, [A] is the molar concentration of the analyte, and Ksv is the quenching constant (M−1), was utilized to evaluate the fluorescence quenching efficiency so as to compare the efficiency of the different MOFs (Fig. 7b). At very low concentrations of TNP, a linear increase in the SV plot was observed, which diverged from linearity and began to bend upwards upon further increasing the concentration. This non-linear nature of the SV plots for TNP suggests the probable mechanism of a combination of static and dynamic quenching processes or an energy transfer phenomenon between TNP and MOFs.19c–e Based on the equation, the quenching constants (Ksv) of MOFs 1–6 for TNP can be estimated to be 8.5 × 104 M−1, 1.8 × 105 M−1, 1.3 × 105 M−1, 2.2 × 105 M−1, 1.2 × 105 M−1, and 1.6 × 105 M−1, respectively (Fig. S18†). However, all the quenching constants are larger than those of a previously reported pyrazine-based MOF sensor.12 As mentioned above, the pyrazine-based MOFs show highly selective fluorescence quenching towards NACs, especially TNP.
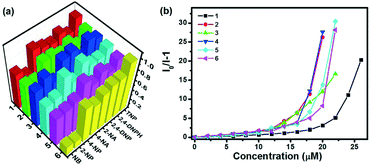 |
| | Fig. 7 (a) Fluorescence quenching efficiencies of MOFs 1–6 (0.5 mg in 2 mL H2O) towards various NACs (30 μM); (b) Stern–Volmer (SV) plots of MOFs 1–6 (0.5 mg in 2 mL H2O) for TNP. | |
Electrocatalytic performances toward the HER
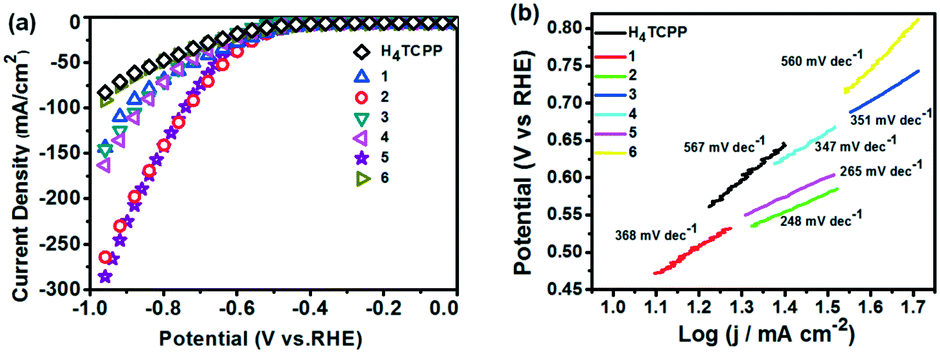
The HER catalytic activities of H4TCPP and 1–6 were evaluated by LSV reaching negative potentials down to −1 V vs. Ag/AgCl.20 These experiments were conducted in 1 M KOH aqueous solution (pH = 14) using an RDE rotating at 1200 rpm in a standard three-electrode mode with a scan rate of 5 mV s−1. The catalyst-based working electrode was obtained through loading the as-prepared samples onto the surface of the GCE.21 The polarization curves of the as-prepared catalyst were obtained by LSV measurements and presented in Fig. 8a. As expected, the electrocatalytic activities of the tested MOFs were obviously improved compared to that of ligand H4TCPP. In particular, 2 and 5 presented markedly enhanced electrocatalytic performances with onset overpotentials of 362 and 244 mV, and achieved a current density of 10 mA cm−2 (j10) at overpotentials of 449 mV and 457 mV, respectively. The corresponding data of H4TCPP and 1–6 are listed in Table S3.† To further demonstrate the stability of MOFs under test conditions, 2 and 5 were chosen to be immersed in 1 M KOH aqueous solution at different times and the corresponding PXRD patterns were recorded. As shown in Fig. S19 and S20,† the PXRD profiles of 2 and 5 were almost unaltered after 4 hours of immersion, revealing the retained crystallinity, which suggested the stability of MOFs under test conditions. The HER kinetics of the tested samples were obtained using the corresponding Tafel plots via fitting the linear regions to the Tafel equation (η = b logj + α, η for the overpotential, b for the Tafel slope, j for the current density, and α for the Tafel constant). Particularly, the Tafel slopes of 2 and 5 were obtained to be ca. 248 and 265 mV dec−1, respectively, which were much smaller than that of the H4TCPP (567 mV dec−1), suggesting more favorable kinetics towards the electrocatalytic HER (Fig. 8b). Subsequently, electrochemical impedance spectroscopy (EIS) was employed to investigate the electrode kinetics under the catalytic HER operating conditions. The Nyquist plots (Fig. S21†) revealed the obviously reduced charge transfer resistance of 2 and 5 in comparison with that of other MOFs and ligand H4TCPP, which also suggested a faster electron transfer process and facile kinetics toward the hydrogen evolution involving the catalyst 2 or 5.
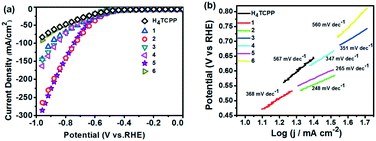 |
| | Fig. 8 (a) HER polarization curves of H4TCPP and compounds 1–6 in 1 M KOH solution with a scan rate of 5 mV s−1. (b) Tafel plots of H4TCPP and compounds 1–6. | |
Conclusions
In summary, we herein presented a series of new MOFs with 3D porous networks based on the backbone of tetracarboxylic acid derived from pyrazine. All MOFs were characterized by single-crystal X-ray diffraction, powder X-ray diffraction, elemental analysis, IR spectroscopy, thermal-gravimetric analysis and fluorescence spectroscopy. In addition, almost all MOFs exhibited sensitive response to NACs, especially to TNP, with fluorescence quenching efficiency behaviors. Remarkably, MOFs 2 and 5 exhibited a certain activity on the electrocatalytic HER without conductive additives or other binders with Tafel slopes of 248 and 265 mV dec−1, respectively. The results indicate that neat MOFs exhibit promising electrocatalytic performances in the HER, which may provide a new avenue for exploring MOF materials in electrocatalytic materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21671159), the Meritocracy Research Funds of China West Normal University (17YC030) and the Key Laboratory of Chemical Synthesis and Pollution Control of Sichuan Province (CSPC201802).
Notes and references
-
(a) P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232 CrossRef CAS PubMed
 ;
(b) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed ;
(c) J. Pang, S. Yuan, J. Qin, C. Liu, C. Lollar, M. Wu, D. Yuan, H. C. Zhou and M. C. Hong, J. Am. Chem. Soc., 2017, 139, 16939 CrossRef CAS PubMed ;
(d) H. L. Wang, Q. L. Zhu, R. Zou and Q. Xu, Chem, 2017, 2, 52 CrossRef CAS ;
(e) X. Gu and D. Xue, CrystEngComm, 2007, 9, 471 RSC .
;
(b) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed ;
(c) J. Pang, S. Yuan, J. Qin, C. Liu, C. Lollar, M. Wu, D. Yuan, H. C. Zhou and M. C. Hong, J. Am. Chem. Soc., 2017, 139, 16939 CrossRef CAS PubMed ;
(d) H. L. Wang, Q. L. Zhu, R. Zou and Q. Xu, Chem, 2017, 2, 52 CrossRef CAS ;
(e) X. Gu and D. Xue, CrystEngComm, 2007, 9, 471 RSC .
-
(a) K. M. L. Taylor-Pashow, J. Della Rocca, Z. Xie, S. Tran and W. B. Lin, J. Am. Chem. Soc., 2009, 131, 14261 CrossRef CAS PubMed ;
(b) J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869 CrossRef CAS PubMed ;
(c) W. J. Ma, Q. Jiang, P. Yu, L. F. Yang and L. Q. Mao, Anal. Chem., 2013, 85, 7550 CrossRef CAS PubMed ;
(d) T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982 RSC ;
(e) Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC ;
(f) S. S. Nagarkar, A. V. Desai and S. K. Ghosh, CrystEngComm, 2016, 18, 2994 RSC ;
(g) M. X. Wu and Y. W. Yang, Adv. Mater., 2017, 29, 1606134 CrossRef PubMed ;
(h) K. Tanaka, K. Sakuragi, H. Ozaki and Y. Takada, Chem. Commun., 2018, 54, 6328 RSC .
-
(a) N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS PubMed ;
(b) M. Balat and H. Balat, Energy Sources, Part A, 2009, 31, 1280 CrossRef CAS ;
(c) T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474 CrossRef CAS PubMed ;
(d) D. Merki and X. Hu, Energy Environ. Sci., 2011, 4, 3878 RSC .
-
(a) M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332 CrossRef CAS PubMed ;
(b) J. A. Turner, Science, 2004, 305, 972 CrossRef CAS PubMed .
-
(a) P. Xiao, M. A. Sk, L. Thia, X. Ge, R. J. Lim, J. Y. Wang, K. H. Lim and X. Wang, Energy Environ. Sci., 2014, 7, 2624 RSC ;
(b) J. Tian, N. Cheng, Q. Liu, W. Xing and X. Sun, Angew. Chem., Int. Ed., 2015, 54, 5493 CrossRef CAS PubMed ;
(c) M. R. Gao, J. X. Liang, Y. R. Zheng, Y. F. Xu, J. Jiang, Q. Gao, J. Li and S. H. Yu, Nat. Commun., 2015, 6, 5982 CrossRef CAS PubMed ;
(d) W. Salomon, G. Paille, M. Gomez-Mingot, P. Mialane, J. Marrot, C. Roch-Marchal, G. Nocton, C. Mellot-Draznieks, M. Fontecave and A. Dolbecq, Cryst. Growth Des., 2017, 17, 1600 CrossRef CAS .
- H. Kita, J. Electrochem. Soc., 1966, 113, 1095 CrossRef CAS .
-
(a) R. Kitaura, S. Kitagawa, Y. Kubota, T. C. Kobayashi, K. Kindo, Y. Mita, A. Matsuo, M. Kobayashi, H. C. Chang, T. C. Ozawa, M. Suzuki, M. Sakata and M. Takata, Science, 2002, 298, 2358 CrossRef CAS PubMed ;
(b) S. Yang, X. Lin, A. J. Blake, G. S. Walker, P. Hubberstey, N. R. Champness and M. Schröder, Nat. Chem., 2009, 1, 487 CrossRef CAS PubMed ;
(c) Y. He, W. Zhou, G. Qian and B. Chen, Chem. Soc. Rev., 2014, 43, 5657 RSC ;
(d) J. Mehta, N. Bhardwaj, S. K. Bhardwaj, K. H. Kim and A. Deep, Coord. Chem. Rev., 2016, 322, 30 CrossRef CAS .
-
(a) A. Mahmood, W. Guo, H. Tabassum and R. Zou, Adv. Energy Mater., 2016, 6, 1600423 CrossRef ;
(b) H. M. Barkholtz and D. J. Liu, Mater. Horiz., 2017, 4, 20 RSC ;
(c) K. Shen, X. Chen, J. Chen and Y. Li, ACS Catal., 2016, 6, 5887 CrossRef CAS ;
(d) Z. Song, N. Cheng, A. Lushington and X. Sun, Catalysts, 2016, 6, 116 CrossRef ;
(e) H. Zhang, H. Osgood, X. Xie, Y. Shao and G. Wu, Nano Energy, 2017, 31, 331 CrossRef CAS .
- D. Sheberla, J. C. Bachman, J. S. Elias, C. J. Sun, Y. Shao-Horn and M. Dincă, Nat. Mater., 2017, 16, 220 CrossRef CAS PubMed .
-
(a) S. S. Nagarkar, B. Joarder, A. K. Chaudhari, S. Mukherjee and S. K. Ghosh, Angew. Chem., 2013, 125, 2953 CrossRef ;
(b) C. Zhang, Y. Yan, L. Sun, Z. Liang and J. Li, CrystEngComm, 2016, 18, 4102 RSC ;
(c) L. Zhang, Z. Kang, X. Xin and D. Sun, CrystEngComm, 2016, 18, 193 RSC ;
(d) Y. T. Yan, J. Liu, G. P. Yang, F. Zhang, Y. K. Fan, W. Y. Zhang and Y. Y. Wang, CrystEngComm, 2018, 20, 477 RSC .
-
(a) W. Y. Guan, F. Xiong, H. L. Zhang, W. Tang, S. F. Zhang, L. H. Jing and D. B. Qin, CrystEngComm, 2014, 16, 7701 RSC ;
(b) S. F. Zhang, F. Xiong, Z. He, Y. Liang, J. R. Xue, L. H. Jing and D. B. Qin, Polyhedron, 2015, 102, 401 CrossRef CAS ;
(c) Y. Liang, W. G. Yuan, S. F. Zhang, Z. He, J. R. Xue, X. Zhang, L. H. Jing and D. B. Qin, Dalton Trans., 2016, 45, 1382 RSC .
- Y. Jiang, L. Sun, J. Du, Y. Liu, H. Shi, Z. Liang and J. Li, Cryst. Growth Des., 2017, 17, 2090 CrossRef CAS .
-
(a)
G. M. Sheldrick, SHELXS 97, Program for Crystal Structure Solution, University of Göttingen, Göttingen, Germany, 1997 Search PubMed ;
(b)
G. M. Sheldrick, SHELXL 97, Program for Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed .
- V. A. Blatov, A. P. Shevchenko and V. N. Serezhkin, J. Appl. Crystallogr., 2000, 33, 1193 CrossRef CAS .
-
(a) D. Singh and C. M. Nagaraja, Cryst. Growth Des., 2015, 15, 3356 CrossRef CAS ;
(b) Z. Ju, W. Yan, X. Gao, Z. Shi, T. Wang and H. Zheng, Cryst. Growth Des., 2016, 16, 2496 CrossRef CAS .
-
(a) J. S. Hu, Y. J. Shang, X. Q. Yao, L. Qin, Y. Z. Li, Z. J. Guo, H. G. Zheng and Z. L. Xue, Cryst. Growth Des., 2010, 10, 4135 CrossRef CAS ;
(b) Z. L. Fang, R. M. Yu, J. G. He, Q. S. Zhang, Z. G. Zhao and C. Z. Lu, Inorg. Chem., 2009, 48, 7691 CrossRef CAS PubMed .
-
(a) Z. Z. Lin, F. L. Jiang, L. Chen, C. Y. Yue, D. Q. Yuan, A. J. Lan and M. C. Hong, Cryst. Growth Des., 2007, 7, 1712 CrossRef CAS ;
(b) P. Cui, Z. Chen, D. L. Gao, B. Zhao, W. Shi and P. Cheng, Cryst. Growth Des., 2010, 10, 4370 CrossRef CAS ;
(c) X. J. Li, Z. J. Yu, T. Guan, X. X. Li, G. C. Ma and X. F. Guo, Cryst. Growth Des., 2015, 15, 278 CrossRef CAS .
- A. Wozna and A. Kapturkiewicz, Phys. Chem. Chem. Phys., 2015, 17, 30468 RSC .
-
(a) M. Wang, V. Vajpayee, S. Shanmugaraju, Y. R. Zheng, Z. Zhao, H. Kim, P. S. Mukherjee, K. W. Chi and P. J. Stang, Inorg. Chem., 2011, 50, 1506 CrossRef CAS PubMed ;
(b) S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 17310 CrossRef CAS PubMed ;
(c) S. S. Nagarkar, A. V. Desai and S. K. Ghosh, Chem. Commun., 2014, 50, 8915 RSC ;
(d) B. Joarder, A. V. Desai, P. Samanta, S. Mukherjee and S. K. Ghosh, Chem. – Eur. J., 2015, 21, 965 CrossRef CAS PubMed ;
(e) S. Khatua, S. Goswami, S. Biswas, K. Tomar, H. S. Jena and S. Konar, Chem. Mater., 2015, 27, 5349 CrossRef CAS .
- Z. Xiao, Y. Wang, S. Zhang, W. Fan, X. Xin, X. Pan, L. Zhang and D. Sun, Cryst. Growth Des., 2017, 17, 4084 CrossRef CAS .
-
(a) Y. P. He, Y. X. Tan and J. Zhang, Cryst. Growth Des., 2013, 13, 6 CrossRef CAS ;
(b) L. Wang, Y. Wu, R. Cao, L. Ren, M. Chen, X. Feng, J. Zhou and B. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 16736 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. CCDC reference numbers 1845900–1845904 and 1846960 for compounds 1–6. Crystal data, additional figures, TGA, PXRD, IR spectra, and optical spectra. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ce01868e |
|
| This journal is © The Royal Society of Chemistry 2019 |