
Mechanochemistry vs. solution growth: striking differences in bench stability of a cimetidine salt based on a synthetic method†
Ghada
Ayoub
a,
Vjekoslav
Štrukil
 *b,
László
Fábián
c,
Cristina
Mottillo
a,
Huizhi
Bao
a,
Yasujiro
Murata
d,
Audrey
Moores
a,
Davor
Margetić
b,
Mirjana
Eckert-Maksić
b and
Tomislav
Friščić
*ab
*b,
László
Fábián
c,
Cristina
Mottillo
a,
Huizhi
Bao
a,
Yasujiro
Murata
d,
Audrey
Moores
a,
Davor
Margetić
b,
Mirjana
Eckert-Maksić
b and
Tomislav
Friščić
*ab
aDepartment of Chemistry, McGill University, Montreal, Quebec, H3A 0B8, Canada. E-mail: tomislav.friscic@mcgill.ca
bRuđer Bošković Institute, Bijenička cesta 54, Zagreb, HR-10000, Croatia. E-mail: vstrukil@irb.hr
cSchool of Pharmacy, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, UK
dInstitute for Chemical Research, Kyoto University, Uji, Kyoto 611-0011, Japan
First published on 25th October 2018
Abstract
A mechanochemically prepared solvated salt of the archetypal blockbuster drug cimetidine exhibits significantly different bench stability to an analogous material made in solution. Samples obtained from solution are stable for weeks at room temperature and 45 °C, but mechanochemically made ones readily desolvate and convert to a new polymorph of non-solvated salt. While mechanochemistry becomes increasingly popular in synthesising drug solid forms, this work illustrates that it can have a profound effect on material stability.
The discovery and preparation of new solid forms of active pharmaceutical ingredients (APIs)1 is an important challenge of modern pharmaceutical materials science, with implications for improving the physicochemical properties of drugs (e.g. solubility,2 bioavailability,3 compressibility,4 dissolution rate,5 taste,6 and colour7) and establishing new intellectual properties.8
Mechanochemical9 techniques, e.g. liquid-assisted grinding (LAG) or polymer-assisted grinding (POLAG), have become of high interest in the API solid form discovery and, recently, API synthesis.10–13 This interest rests on their short reaction times and ability to circumvent limitations of solubility, solvolysis or thermal degradation.14–17 Particularly notable is LAG, which uses a catalytic amount of a liquid to accelerate mechanochemical reactions and direct formation of polymorphs or stoichiometric variations of cocrystals or salts.1,18–23 While LAG reactions of molecular crystals are rapid, often enabling complete conversion in minutes,24 they are also scalable to gram amounts in the laboratory.25 In the context of scale-up and manufacturing, twin screw extrusion now permits continuous mechanosynthesis of pharmaceutical cocrystals and organic molecules.26 As mechanochemistry becomes increasingly significant in pharmaceutical materials science, most reports have focused on its efficiency in solid form synthesis and discovery. In contrast, little or no attention has been paid to validation of mechanochemical products, by identifying potential differences in their physicochemical properties compared to those of nominally identical materials obtained by solution techniques.
We now highlight the need for such critical validation of mechanochemically made materials by describing stark differences in the bench stability of a mechanochemically prepared solvate of a salt of the API cimetidine27 (cim) and fumaric acid (H2fum) (Fig. 1a), compared to that of an analogous material made from solution.
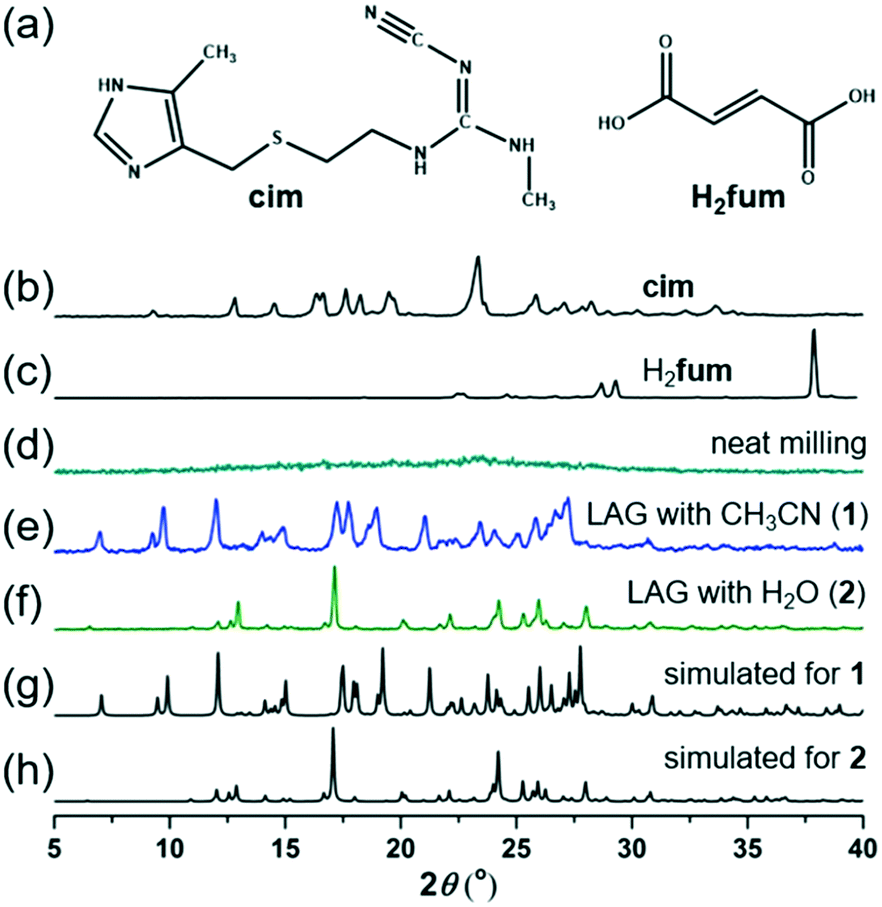 | ||
| Fig. 1 (a) Structures of cim and H2fum. PXRD patterns of: (b) commercial cim and (c) H2fum; (d) the product of neat grinding of cim and H2fum; (e) 1, the product of LAG with MeCN (η = 0.30 μL mg−1);20 and (f) 2, the product of LAG with water (η = 0.30 μL mg−1).20 Simulated PXRD patterns for (g) the crystal structure of 1 and (h) the crystal structure of 2. | ||
Cimetidine (cim) is a well-known histamine H2-receptor antagonist. Marketed as Tagamet, solid cim is of outstanding importance in pharmaceutical materials science being the first drug to reach $1 billion in annual sales.27,28 It remains widely used in heartburn and peptic ulcer treatment, with recent work also indicating anti-cancer activity.29 In contrast to its status as the pioneering beta-blocker and the archetypal blockbuster drug, there have been no reports on crystal engineering of solid forms of cim, with existing structural studies dealing with polymorphs, the hydrochloride salt and metal complexes.30
Neat milling of cim and H2fum in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio led to amorphization, as shown by a featureless powder X-ray diffraction (PXRD) pattern (Fig. 1). However, 20 minutes of LAG with water or acetonitrile (MeCN) led to the disappearance of reactant X-ray reflections and the formation of new crystalline products. The use of MeCN gave 1, characterized by Bragg reflections distinct from those of any known forms of cim and H2fum (Fig. 1).30,31 In contrast, the use of water gave a material (2) with a PXRD pattern different from that of 1 or any reported forms of cim and H2fum (Fig. 1).
:1 stoichiometric ratio led to amorphization, as shown by a featureless powder X-ray diffraction (PXRD) pattern (Fig. 1). However, 20 minutes of LAG with water or acetonitrile (MeCN) led to the disappearance of reactant X-ray reflections and the formation of new crystalline products. The use of MeCN gave 1, characterized by Bragg reflections distinct from those of any known forms of cim and H2fum (Fig. 1).30,31 In contrast, the use of water gave a material (2) with a PXRD pattern different from that of 1 or any reported forms of cim and H2fum (Fig. 1).
The crystal structure of 1 was determined by X-ray diffraction on crystals obtained by re-crystallisation of the mechanochemical product from MeCN (Fig. 2). Compound 1 is a solvated salt in which the asymmetric unit consists of singly protonated cimH+ cations, Hfum− anions, and MeCN molecules disordered on a crystallographic inversion centre. Structural analysis and 1H nuclear magnetic resonance (NMR) spectroscopy in DMSO-d6 revealed that 1 contains cim, H2fum and MeCN in a 1:1:0.5 respective ratio, consistent with the formula (cimH+)(Hfum−)·0.5MeCN. The structure is composed of cyclic hydrogen-bonded dimers of cimH+, held together by two N–H⋯N hydrogen bonds. The cationic dimers associate by N–H⋯O hydrogen bonds to chains of hydrogen-bonded Hfum− anions that propagate along the crystallographic b-direction, forming a three-dimensional hydrogen-bonded structure. The ionic nature of 1 is confirmed by the carbon–oxygen bond (C–O bond) lengths in Hfum− anions. One of the carboxylate moieties on each anion exhibited very similar C–O bond distances of 1.246(4) Å and 1.274(3) Å, consistent with a deprotonated carboxylate group, while the other one exhibited significantly shorter 1.212(4) Å and one longer 1.308(4) Å C–O bond, consistent with a neutral acid group. The acetonitrile molecule in the structure of 1 does not appear to be involved in any significant intermolecular interactions, except a potential C–H⋯N hydrogen bonding interaction (C⋯N separation 3.659(5) Å)32 between the nitrogen atom of the MeCN molecule and the methylene moiety in the 4-position of the imidazole ring of cimetidine (see the ESI†).
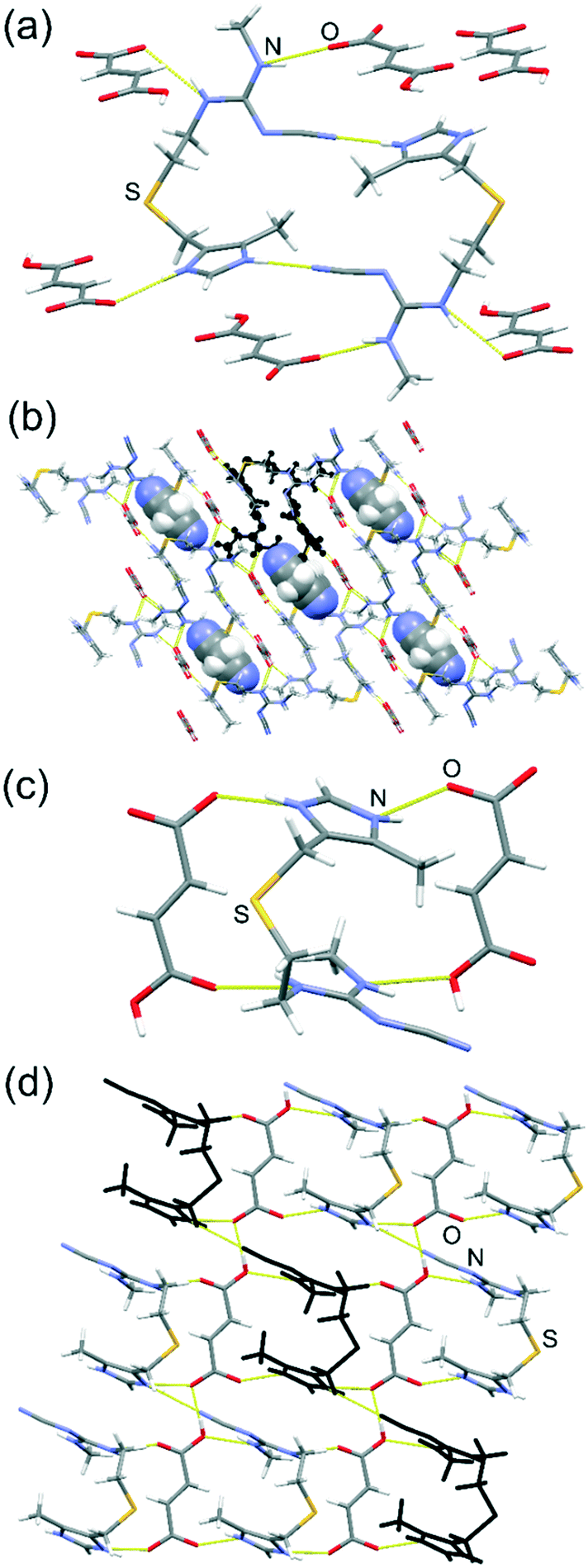 | ||
| Fig. 2 Crystal structures of 1 and 2: (a) a single hydrogen-bonded dimer of cimH+ in 1, associated with surrounding Hfum− anions by N–H⋯O hydrogen bonds; (b) structure of 1 viewed along the crystallographic b-axis, with disordered acetonitrile guests shown in a space-filling model, and one of the hydrogen-bonded cimH+ dimers highlighted in black; (c) view of a single cimH+ cation bridging neighboring Hfum− anions via N–H⋯O bonds and (d) a single hydrogen-bonded layer in the structure of 2 viewed along the crystallographic c-axis, with a chain of hydrogen-bonded cimH+ highlighted in black. | ||
Attempts to obtain diffraction-quality crystals of 2 were unsuccessful, requiring structure characterisation from PXRD data (see the ESI†). Indexing of the PXRD pattern of 2, after conversion to the conventional reduced cell, revealed a triclinic structure with a = 7.8985 Å, b = 8.3479 Å, c = 14.2018 Å, α = 87.575°, β = 75.273°, γ = 76.159° and a volume of 879.2 Å3. The structure solution revealed that 2 is a non-solvated salt with composition (cimH+)(Hfum−), with the ionic nature of 1 and 2 being confirmed by the natural abundance 15N CP-MAS solid-state NMR spectra, which resemble those of cim hydrochloride, but are different from those of neutral cim (see the ESI†). The absence of solvent in 2 was also confirmed by thermogravimetric analysis (TGA, see the ESI†). The structure of 2 consists of layers in the crystallographic (001) plane, in which cimH+ cations bridge parallel chains of hydrogen-bonded Hfum− anions via N–H⋯O hydrogen bonds. The cimH+ cations in each layer form chains held by N–H⋯N hydrogen bonds, propagating in the crystallographic [110]-direction (Fig. 2c and d).
While samples of 1 prepared mechanochemically and from solution are nominally identical, they exhibit very different bench stabilities. Exposure of mechanochemically made 1 to 45 °C over two days led to the disappearance of the prominent (10−1) X-ray reflection at 2θ = 7.0° (Fig. 3). Identical behavior was observed regardless of the amount of acetonitrile used in the LAG synthesis of 1, as demonstrated by samples prepared at liquid-to-solid η20 ratios of 0.15, 0.30, 0.45 and 0.60 μL mg−1 (see the ESI†).
 | ||
| Fig. 3 Analysis of 1 before and after two days at 45 °C in air. PXRD patterns of: (a) the mechanochemically made sample and (b) the mechanochemically made sample after exposure to 45 °C. Simulated PXRD patterns of (c) 1 and (d) 1′. Section of the 1H-NMR spectrum recorded in DMSO-d6 for (e) solution-made 1 before (top) and after thermal treatment (bottom) and (f) mechanochemically made 1 before (top) and after exposure to 45 °C (bottom). SEM images of mechanochemically made 1 (g) before and (h) after exposure to 45 °C. Scale bar corresponds to 1 μm. | ||
The remainder of the PXRD pattern changed less significantly, suggesting that the product (1′) is structurally similar to 1. The 1H-NMR analysis of a solution of 1′ in DMSO-d6 revealed the absence of MeCN and the composition (cimH+)(Hfum−), identical to 2. Scanning electron microscopy (SEM) showed that mechanochemically made 1 consisted of elongated cuboid particles with a length of 228 ± 85 nm (Fig. 3g). After thermal desolvation, little change in particle size or morphology was observed, consistent with retention of crystallinity (Fig. 3g and h).
Indexing of PXRD data for 1′ revealed a monoclinic unit cell strongly resembling that of 1, with a = 13.770(1) Å, b = 8.0432(5) Å, c = 18.949(1) Å, β = 107.419(4)° and V = 2002.48(21) Å3. The simulated annealing structure solution and Rietveld refinement in the space group P21/n confirmed that 1′ is indeed a polymorph of the non-solvated salt 2, isostructural to 1 (Fig. 4a, also see the ESI†). The absence of solvent in 1′ was also verified by TGA (see the ESI). The crystal structure analysis readily explains the significant reduction in the intensity of the (10−1)-reflection upon desolvation of 1 into 1′: the (10−1)-planes of 1 are populated with guest MeCN molecules, and their removal leads to formation of voids illustrated in Fig. 4b and a reduction in electron density contributing to X-ray scattering from those crystal planes.
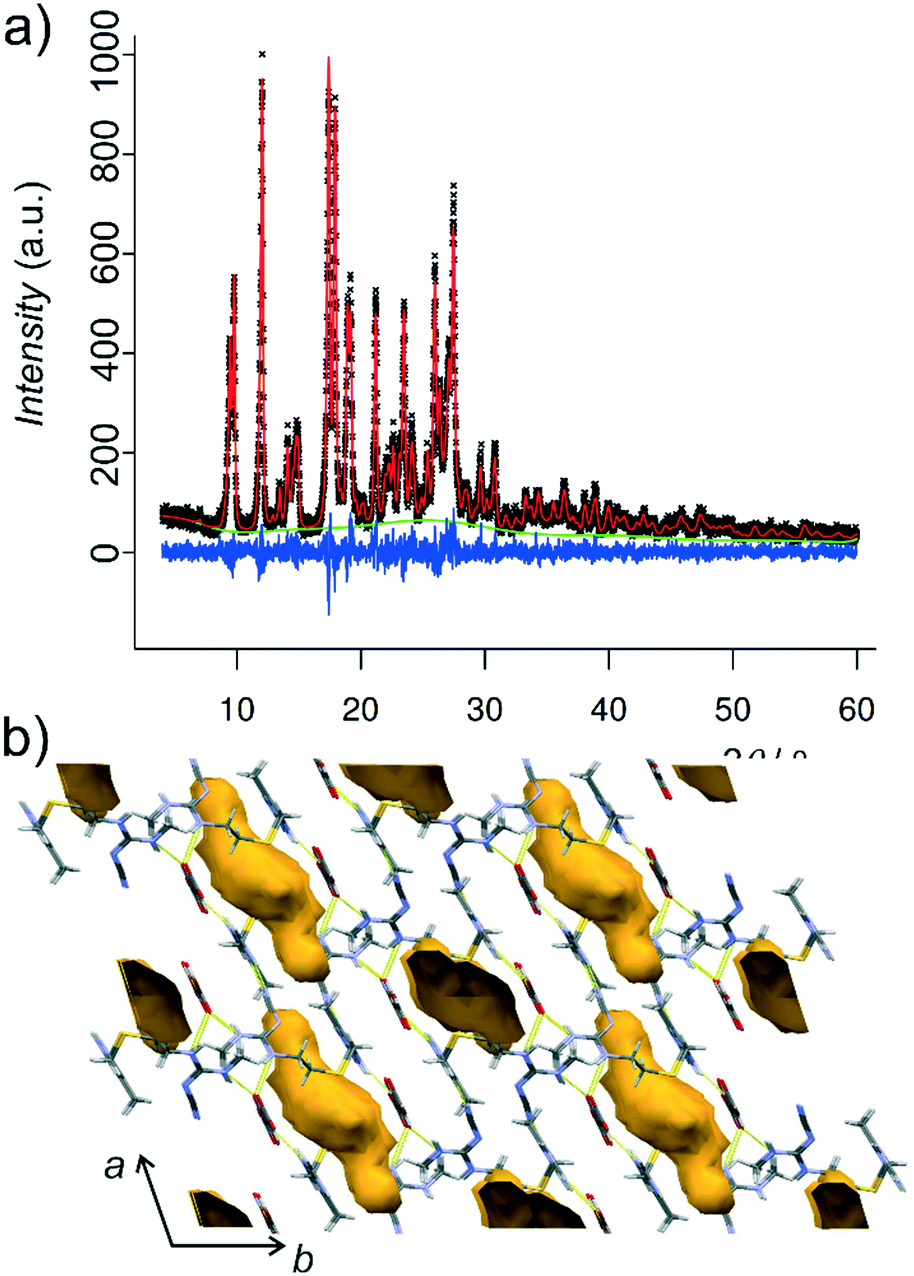 | ||
| Fig. 4 a) Final Rietveld fit for the structure of 1′ determined from the PXRD data and b) the crystal structure of 1′ viewed parallel to the crystallographic b-axis and displaying the voids (yellow, detected using a spherical probe of 1.2 Å in radius) previously occupied by MeCN guests. | ||
In contrast, solution-made 1 was significantly more resistant to thermal treatment. The crystal structure analysis of a single crystal of 1, after being kept at 45 °C for two days, revealed no evidence of acetonitrile loss. This was corroborated by solution 1H-NMR analysis of thermally treated crystals, which revealed a loss of less than 10% of the initial amount of MeCN. Similarly, no significant difference was observed in the crystallographic unit cell parameters or the overall appearance of X-ray diffraction spots of 1 upon exposure to 45 °C over a period of 5 and 10 days (see the ESI†) (Table 1).
| t/days | a/Å | b/Å | c/Å | β/° |
|---|---|---|---|---|
| 0 | 13.804(1) | 8.0191(7) | 18.715(2) | 107.580(3) |
| 5 | 13.799(1) | 8.0172(7) | 18.723(2) | 107.657(3) |
| 10 | 13.792(1) | 8.0148(7) | 18.726(2) | 107.555(3) |
The striking difference in the stability of mechanochemically- and solution-made 1 is even more evident from 1H-NMR monitoring of the MeCN content in samples exposed to air at room temperature (Fig. 5a and b). Mechanochemically made 1 lost almost all MeCN within 40 h, while solution-grown crystals remained solvated even after 15 days. Indeed, complete removal of MeCN from solution-grown 1 was difficult even upon harsher treatment: after exposure to 80 °C and reduced pressure of 0.2 bar for 10 days, 1H-NMR analysis still revealed the presence of 0.15 molecules of MeCN per (cimH+)(Hfum−) unit. The most likely explanation for the observed stability differences is the particle size. As revealed by SEM, solution-grown crystals of 1 are much larger than mechanochemically made ones, appearing as needles with a length on the order of 1 mm (Fig. 6). To qualitatively evaluate the effect of the crystal size, we studied the effect of mechanical treatment on solution-grown 1, by either gentle or vigorous grinding using a mortar and pestle.
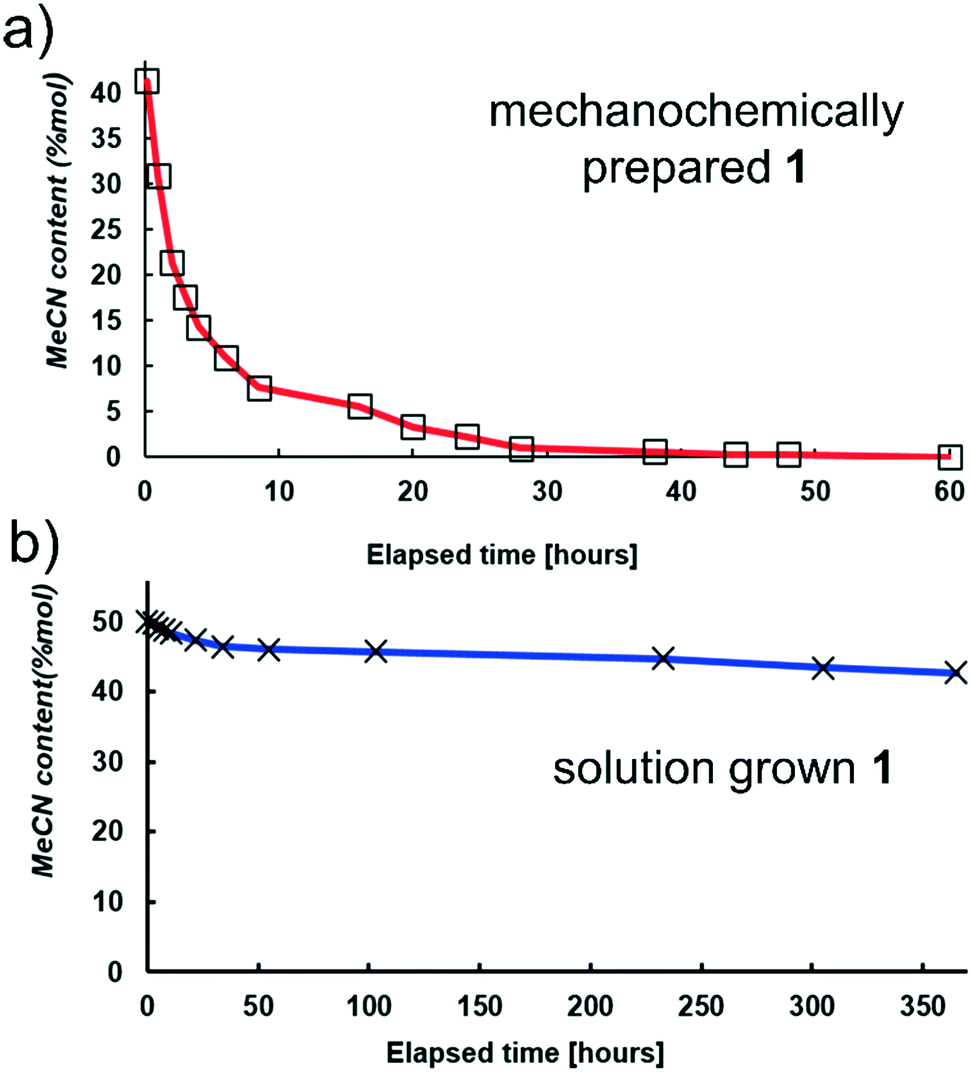 | ||
| Fig. 5 Difference in rates of acetonitrile (MeCN) loss in open air, at room temperature, for samples of 1 that were: a) mechanochemically prepared and b) solution grown. | ||
 | ||
| Fig. 6 SEM images of: (a) solution grown crystals of 1 (scale bar = 400 μm) and solution grown crystals of 1 after (b) gentle grinding (scale bar = 400 μm) and (c) harsher grinding using a mortar and pestle (scale bar = 40 μm). | ||
SEM analysis revealed that gentle grinding fragmented the crystals into smaller particles with an average size of around 230 μm in length, while harsher grinding led to an average size of ca. 19 μm (see the ESI†). After 2 days at 45 °C in open air, the gently ground sample underwent more significant MeCN loss (50% of the original content) compared to unperturbed crystals (2%). The sample produced by harsher grinding lost ca. 78% of the original MeCN content, resulting in a material with composition (cimH+)(Hfum−)·0.11 MeCN. These results, summarized in Table S1 in the ESI,† support the view that the stability differences between mechanochemically and solution-grown 1 are likely due to their different particle sizes and defects (Fig. 6).33,34
In summary, we described a significant difference in the stability between nominally identical solid forms of cimetidine, prepared by mechanochemistry or solution growth. So far, studies of mechanochemical synthesis of API solid forms have focused on screening and quantitative synthesis. However, this work highlights a not yet explored effect33,34 of mechanochemistry on the solid-state properties of solid API forms. While this effect herein led to the discovery of a new polymorph of a not previously described salt solid form of cimetidine, in a wider context it can be regarded as a potential problem when mechanochemical techniques are employed. Consequently, this work highlights a growing need to investigate and validate the properties of mechanochemically made materials with respect to analogous ones made by different methods.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Unity Through Knowledge fund (project no. 63/10), the NSERC Discovery Grant (RGPIN-2017-06467), and the E. W. R. Steacie Memorial Fellowship (SMFSU 507347-17). GA acknowledges the Ludo Frevel Crystallography Scholarship and the McGill University Departmental Fellowship. We thank Prof. J. Bernstein for advice and critical reading of the manuscript and Drs. Fred Morin and Robin S. Stein for acquiring the solid-state NMR data.Notes and references
-
(a) N. K. Duggirala, M. L. Perry, Ö. Almarsson and M. J. Zaworotko, Chem. Commun., 2016, 52, 640 RSC
; (b) D. J. Berry and J. W. Steed, Chem. Commun., 2017, 117, 3 CAS
-
(a) D. P. Elder, R. Holm and H. Lopez de Diego, Int. J. Pharm., 2013, 453, 88 CrossRef CAS PubMed
- D. P. McNamara, S. L. Childs, J. Giordano, A. Iarriccio, J. Cassidy, M. S. Shet, R. Mannion, E. O'Donnell and A. Park, Pharm. Res., 2006, 23, 1888 CrossRef CAS PubMed
-
(a) D.-K. Bučar, J. A. Elliott, M. D. Eddleston, J. K. Cockroft and W. Jones, Angew. Chem., Int. Ed., 2015, 54, 249 CrossRef PubMed
- M. Žegarac, E. Lekšić, P. Šket, J. Plavec, M. D. Bogdanović, D.-K. Bučar, M. Dumić and E. Meštrović, CrystEngComm, 2014, 16, 32 RSC
- A. R. Patel and P. R. Vavia, AAPS PharmSciTech, 2008, 9, 544 CrossRef CAS PubMed
- D.-K. Bučar, S. Filip, M. Arhangelskis, G. O. Lloyd and W. Jones, CrystEngComm, 2013, 15, 6289 RSC
- A. V. Trask, Mol. Pharmaceutics, 2007, 4, 301 CrossRef CAS PubMed
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413 RSC
- S. L. Childs, N. Rodríguez-Hornedo, L. S. Reddy, A. Jayasankar, C. Maheshwari, L. McCausland, R. Shipplett and B. C. Stahly, CrystEngComm, 2008, 10, 856 RSC
- D. Braga, L. Maini and F. Grepioni, Chem. Soc. Rev., 2013, 42, 7638 RSC
-
(a) D. Tan, L. Loots and T. Friščić, Chem. Commun., 2016, 52, 7760 RSC
- Y. Zhou, F. Guo, C. E. Hughes, D. L. Browne, T. R. Peskett and K. D. M. Harris, Cryst. Growth Des., 2015, 15, 2901 CrossRef CAS
- D. Hasa, G. Schneider Rauber, D. Voinovich and W. Jones, Angew. Chem., Int. Ed., 2015, 54, 7371 CrossRef CAS PubMed
- D.-K. Bučar, J. A. Elliott, M. D. Eddleston, J. K. Cockcroft and W. Jones, Angew. Chem., Int. Ed., 2015, 54, 249 CrossRef PubMed
- E. Lu, N. Rodríguez-Hornedo and R. Suryanarayanan, CrystEngComm, 2008, 10, 665 RSC
- D. J. Berry, C. C. Seaton, W. Clegg, R. W. Harrington, S. J. Coles, P. N. Horton, M. B. Hursthouse, R. Storey, W. Jones, T. Friščić and N. Blagden, Cryst. Growth Des., 2008, 8, 1697 CrossRef CAS
- T. Friščić, J. Mater. Chem., 2010, 20, 7599 RSC
- W. Jones and M. D. Eddleston, Faraday Discuss., 2014, 170, 9 RSC
- T. Friščić, S. L. Childs, S. A. A. Rizvi and W. Jones, CrystEngComm, 2009, 11, 418 RSC
- A. V. Trask, W. D. S. Motherwell and W. Jones, Chem. Commun., 2004, 890 RSC
- T. Friščić, A. V. Trask, W. Jones and W. D. S. Motherwell, Angew. Chem., Int. Ed., 2006, 45, 7546 CrossRef PubMed
- R. Banerjee, P. M. Bhatt, N. V. Ravindra and G. R. Desiraju, Cryst. Growth Des., 2005, 5, 2299 CrossRef CAS
- K. L. Nguyen, T. Friščić, G. M. Day, L. F. Gladden and W. Jones, Nat. Mater., 2007, 6, 206 CrossRef CAS PubMed
- X. Ma, G. K. Lim, K. D. M. Harris, D. C. Apperley, P. N. Horton, M. B. Hursthouse and S. L. James, Cryst. Growth Des., 2012, 12, 5869 CrossRef CAS
- D. Daurio, C. Medina, R. Saw, K. Nagapudi and F. Alvarez-Núñez, Pharmaceutics, 2011, 3, 582 CrossRef CAS PubMed
- R. W. Brimblecombe, W. A. M. Duncan, G. J. Durant, J. C. Emmett, C. R. Ganellin, G. B. Leslie and M. E. Parsons, Gastroenterology, 1978, 74, 339 CAS
- M. Freemantle, Chem. Eng. News, 2005, 83, 118 CrossRef
-
(a) Y. Zheng, M. Xu, X. Li, J. Jia, K. Fan and G. Lai, Mol. Immunol., 2013, 54, 74 CrossRef CAS PubMed
-
(a) D. A. Middleton, C. S. Le Duff, X. Peng, D. G. Reid and D. Saunders, J. Am. Chem. Soc., 2000, 122, 1161 CrossRef CAS
- The Cambridge Structural Database (CSD) contains 17 entries involving the cimetidine backbone, including cimetidinium hydrochloride (EHIWEZ), cimetidinium hydrochloride monohydrate (CADVIM), cimetidine monohydrate (CIMGUA), three polymorphs (CIMETD, CIMETD01, CIMETD02, CIMETD03, CIMETD04), five Cu2+ (CMTCUA, CONYUZ, CONYUZ01, DEFWEQ, GEWYUC), two Ni2+ (DOKGEP, GAXVUW), Pt2+ (INOPEG) and Co2+ (DOKGAL) complexes.
- Evaluated using PLATON: A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed
- For a study on how particle size can affect relative stabilities of polymorphs formed by mechanochemistry, see: A. M. Belenguer, G. I. Lampronti, A. J. Cruz-Cabeza, C. A. Hunter and J. K. M. Sanders, Chem. Sci., 2016, 7, 6617 RSC
- For comparison of thermodynamic stability of a material prepared mechanochemically, through accelerated aging and solution techniques, see: Z. Akimbekov, A. D. Katsenis, G. P. Nagabushana, G. Ayoub, M. Arhangelskis, A. J. Morris, T. Friščić and A. Navrotsky, J. Am. Chem. Soc., 2017, 139, 7952 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, TGA, FTIR-ATR, solid-state NMR, and SEM data. CCDC 1862747–1862749. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ce01727a |
| This journal is © The Royal Society of Chemistry 2018 |