
Polyoxometalate-based metal–organic framework loaded with an ultra-low amount of Pt as an efficient electrocatalyst for hydrogen production†
Haiyue
Peng‡
a,
Xiaojian
Yang‡
a,
Yuanyuan
Ma
*ab,
Jianing
Liu
a,
Yonghui
Wang
*a,
Huaqiao
Tan
 a and
Yangguang
Li
*a
a and
Yangguang
Li
*a
aKey Laboratory of Polyoxometalate Science of the Ministry of Education, Faculty of Chemistry, Northeast Normal University, Changchun, 130024, China. E-mail: liyg658@nenu.edu.cn; wangyh319@nenu.edu.cn; mayy334@nenu.edu.cn
bCollege of Chemistry & Material Science, Hebei Normal University, Shijiazhuang, Hebei 050024, China
First published on 13th August 2018
Abstract
The development of highly active and stable electrocatalysts for HER has been received extensive attention. Among these, minimizing the Pt usage and promoting its utilization efficiency are essential prerequisites for a future hydrogen economy. Herein, we use a new ε-Keggin-type polyoxometalate-based metal–organic framework H3[Zn4(bimb)2][PMoV8MoVI4O40]·12H2O (POMOF-1) as a support to load a low amount of ultrasmall Pt nanoparticles (NPs), and then mix this material with Ketjenblack carbon (KB) to form a new composite catalyst (Pt@POMOF-1/KB). This composite catalyst shows a low Pt loading amount of 0.43 wt% and the average size of the Pt NPs is 1.5 nm. Pt@POMOF-1/KB exhibits a remarkable HER performance in 0.5 M H2SO4. It only requires an overpotential to of 23 mV to observe a current density of 10 mA cm−2, which is superior to that of 20% Pt/C. The mass activity of Pt@POMOF-1/KB is almost 100 and 37 times that of 20% Pt/C at overpotentials of 50 and 100 mV, respectively, as a consequence of the very low noble-metal loading. This result may provide an approach to meet the cost requirements for large-scale applications.
1 Introduction
With environmental degradation and the depletion of carbon-based fuels, exploring an environmentally friendly energy source to replace conventional fuels has become more and more pressing.1–3 Hydrogen (H2) is considered as an alternative to fossil fuels due to its high energy density, renewability and environmental-friendliness. To obtain H2, water electrolysis has attracted considerable attention due to its stable output, high energy conversion efficiency and product purity.4–6 Currently, platinum (Pt)-based materials are regarded as the most effective and catalytically stable electrocatalysts for the hydrogen evolution reaction (HER).7,8 However, the high cost and low abundance of Pt oblige us to reduce the dosage of Pt while retaining high HER activity. Thus, exploring facile pathways to achieve this purpose has become highly desirable.Nowadays, researchers have identified two main strategies to allay the exorbitant cost of Pt by either superseding or reducing Pt consumption while realizing sustainable H2 production. Recently, the development of electrocatalysts based on transition metals (such as Fe, Ni, Co, Mo and W and their derivatives) have become universal methods for replacing Pt-based noble metal electrocatalysts.9–15 Although considerable efforts have been made, the cycling stability and poor electronic conductivity of these catalysts still impede their practical application for commercial device fabrication and the future hydrogen economy.16,17 Hence, developing a technology which can remarkably minimize the Pt use and in parallel enhance its exploitation efficiency are key factors for the practical application of hydrogen energy. One strategy is to use supports to disperse Pt nanoparticles (NPs) for minimal yet effective utilization.18–22 Up to now, mostly metal oxide supports have been employed to immobilize Pt catalyst, but such metal oxide supports suffer from low surface area and insufficient interaction with Pt atoms, which hinder their application for catalyzing electrochemical reactions.23–25 An ideal support should possess large surface area and strong interaction with Pt along with good electro-conductivity.
Polyoxometalate-based metal–organic frameworks (POMOFs), combining the large surface area of MOFs and unique advantages of POMs have been regarded as promising supports in many fields, such as biomass detection and catalysis.26–31 Among them, POMs, especially highly reduced POMs, possessing high negative charges, controllable redox potentials and modifiable oxygen-rich surfaces, have been proved to be a potentially promising electrocatalysts toward HER.32 More importantly, these highly reduced POMs show strong interaction with Pt atoms, which make them a promising supports to anchor ultra-small Pt nanoparticles.33–35 The high surface area and abundant porous channels of MOFs can not only realize the high dispersion of POMs, protecting them from leaching, but also can induce a confinement effect to prevent the Pt nanoparticles from overgrowth and aggregation. Thus, the highly reduced POM-based MOFs can be promising carriers for the decrease of Pt usage and promotion of HER activity. Moreover, if these Pt-loaded POMOFs are further combined with electro-conductive carbon materials such as carbon nanotubes (CNTs), graphene or Ketjenblack carbon (KB), the as-prepared composite electrocatalysts will further enhance the electron-conductivity and stability.36–39 Taking into account the above considerations, we have prepared a new type of POMOF based on highly reduced ε-Keggin-type POM units, H3[Zn4(bimb)2][PMoV8MoVI4O40]·12H2O (POMOF-1). Utilizing an adsorption and light-reduction method, ultra-small Pt NPs were loaded on the POMOF-1. Then, Pt@POMOF-1 was mixed with KB to form a final composite material (Pt@POMOF-1/KB) as a new type of HER electrocatalyst. The optimized loading of Pt in Pt@POMOF-1/KB is 0.43 wt% and the average size of Pt nanoparticles is 1.5 nm. In 0.5 M H2SO4, Pt@POMOF-1/KB catalyst requires an overpotential of only 23 mV to reach a current density of 10 mA cm−2, which is superior to that of 20% Pt/C. The mass activity of Pt@POMOF-1/KB is almost 100 and 37 times that of 20% Pt/C at overpotentials of 50 and 100 mV respectively, as a consequence of the very low noble-metal loading.
2 Results and discussion
2.1 Synthesis and crystal structure of POMOF-1
POMOF- 1 was hydrothermally synthesized from an aqueous mixture of [NH4]6[Mo7O24]·4H2O, Mo powder, H3PO4, ZnCl2, 1,4-bis(1-imidazolyl)benzene (bimb) and Bu4N(OH) at 180 °C for 120 h. Single-crystal X-ray diffraction analysis revealed that POMOF-1 crystallizes in the tetragonal crystal system with I41/amd space group. The crystallographically asymmetric unit comprised one ε-Keggin-type POM [Zn4PMoV8MoVI4O40]3− unit and four bimb organic bridging ligands, which are linked together through four capped Zn2+ ions on the POM surface (Fig. S1†). The whole compound was charge-balanced by three extra protons, which were most probably located on lattice water molecules, rather than on the surface O sites of the POM units, based on BVS calculations and the XPS spectrum (Table S1 and Fig. S2†). All the ZnII centers display a tetra-coordinated mode, surrounded by one nitrogen atom originating from one bimb ligand and three oxygen atoms derived from one POM unit (Fig. S1†). The Zn–N/O bond lengths were within the range 1.956(9)–1.99(12) Å, while the N/O–Zn–O/N bond angles were within the range 101.2(5)–115.8(5)° (Table S2†). In POMOF-1, the adjacent [Zn4PMoV8MoVI4O40]3− units are connected by bimb bridging ligands, forming a 3D framework with a diamond topology (Fig. 1). It is worth noting that there are five-interpenetration nets in POMOF-1 (Fig. S3 and S4†). As shown in Fig. 1, the window size of the diamond-like framework units is 36.20 × 25.52 Å, which allow four other identical frameworks to interpenetrate it in a parallel fashion, resulting in 5-fold interpenetrating networks, which is rarely reported in POMOF structures. Notably, this 5-fold interpenetrating diamond-like framework contains 1D channels with a window size of 9.5 × 9.5 Å along the c axis (Fig. 2 and S5–S7†). The solvent-accessible void in POMOF-1 is 41.9% calculated by using the PLATON program.40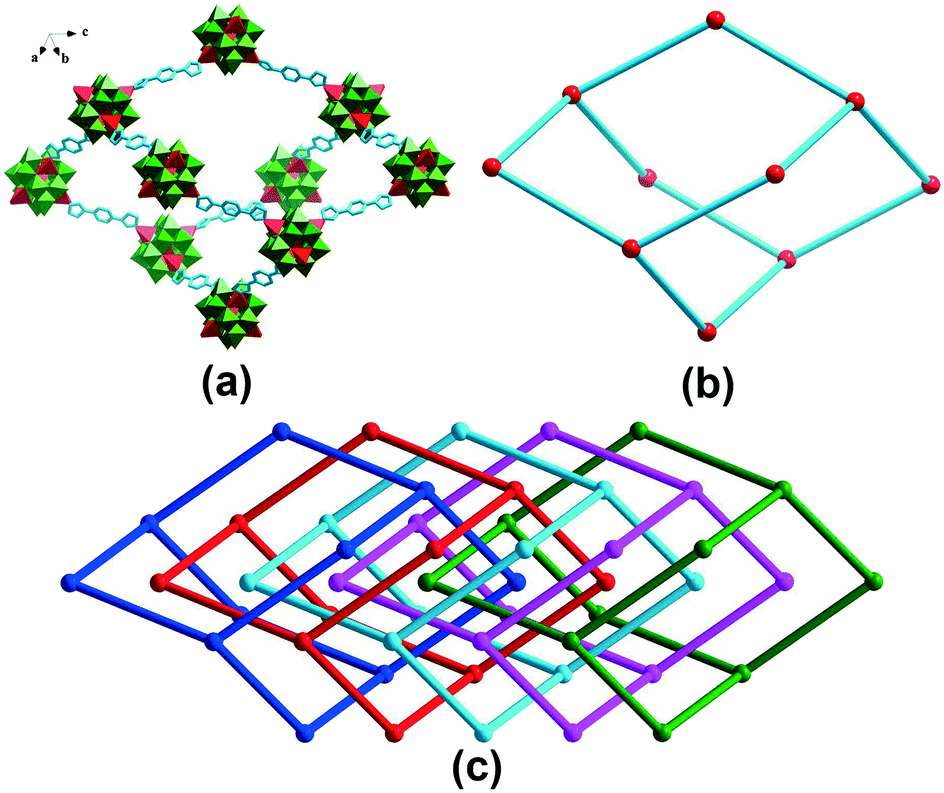 | ||
| Fig. 1 (a) Polyhedral and ball-and-stick representations of the diamond-like fragment in POMOF-1; (b) schematic view of the diamond-like framework in POMOF-1; (c) schematic view of the five-fold interpenetrated diamond-like framework in POMOF-1. | ||
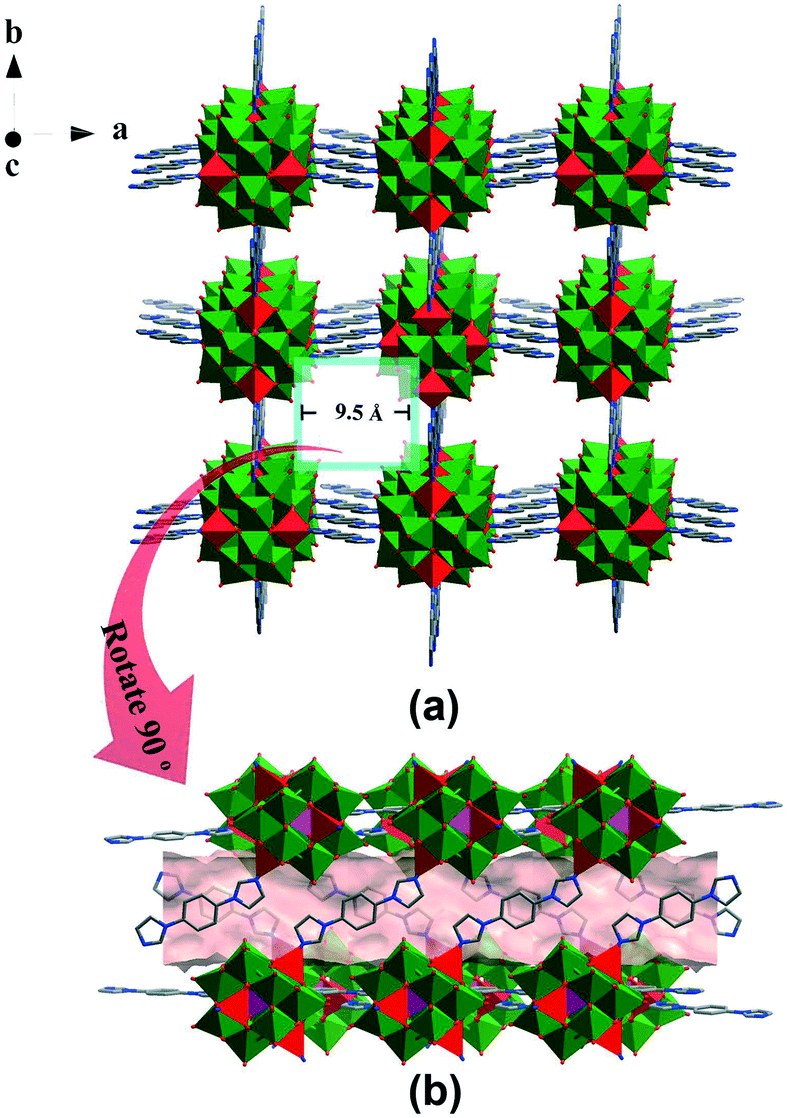 | ||
| Fig. 2 (a) Ball-and-stick and polyhedral view of the 3D diamond-like framework of POMOF-1 with 1D channels viewed along the c axis; (b) the channel along [001] direction in POMOF-1. The solvent accessible voids are modeled with pink background. | ||
Moreover, POMOF-1 exhibits good stability and insolubility in common organic solvents, such as ethanol, methanol and isopropanol (Fig. S8a†). Remarkably, POMOF-1 is also stable in acidic solution in the pH range of 0–7 at room temperature, as confirmed by subsequent XRD measurements (Fig. S8b†). The good solvent and pH stability implies that POMOF-1 can be a good support for further experiments.
2.2 Preparation and characterization of Pt@POMOF-1/KB
Since ε-Keggin-type [Zn4PMoV8MoVI4O40]3− unit is in a highly reduced form, noble metal ions can be easily adsorbed around the highly-reduced POM species and in situ reduced into the corresponding metal nanocomposites upon irradiation.41,42 Combining the advantages of the open framework with a 1D channel system, good stability and highly-reduced POM species, POMOF-1 can be a highly suitable carrier to immobilize noble metal nanoparticles, such as Pt. Considering the electro-conductivity of composite materials as electrocatalysts, Ketjenblack (KB), a highly conductive carbon black, was introduced into the system. Based on the above considerations, Pt@POMOF-1/KB composite material was fabricated by a simple three-step method (Scheme 1). In the typical synthesis process, immersion of as-synthesized POMOF-1 in H2PtCl6 (1 mg ml−1) aqueous solution at ambient temperature afforded Pt-adsorbed POMOF-1 precursor which was then dispersed in isopropanol solution. After illumination with UV light for 30 min, Pt ions were in situ reduced into Pt nanoparticles (NPs) and confined around the POM units, resulting in Pt@POMOF-1. Subsequently, KB and Pt@POMOF-1 were dispersed in ethanol and sonicated for 120 min, generating the final Pt@POMOF-1/KB composite material.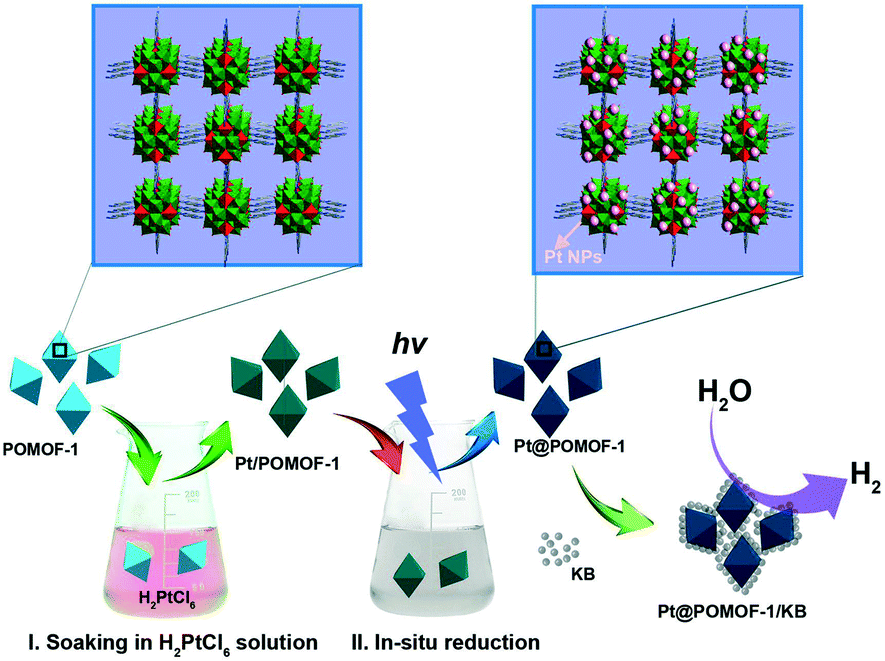 | ||
| Scheme 1 Schematic view of the brief preparation procedure of Pt@POMOF-1/KB composite material. | ||
The morphology and structure of POMOF-1 and Pt@POMOF-1/KB were analyzed by SEM and TEM images (Fig. 3). As shown in Fig. 3a, POMOF-1 has a nano-octahedral shape with average size of 100 nm. The SEM and TEM images demonstrate that Pt@POMOF-1/KB inherits the octahedral morphology of POMOF-1, where the octahedral nanocrystals are surrounded by KB nanospheres (Fig. 3b and c), which enhance the electro-conductivity. High-resolution TEM images of Pt@POMOF-1/KB demonstrate that the Pt particles are uniformly distributed in POMOF-1 and the average size of Pt NPs is ca. 1.5 nm (Fig. 3d and Fig. S9†). EDX element mapping confirms the uniform distribution of Zn, Mo, C, N, O, P and Pt elements in Pt@POMOF-1/KB (Fig. 3e–j).
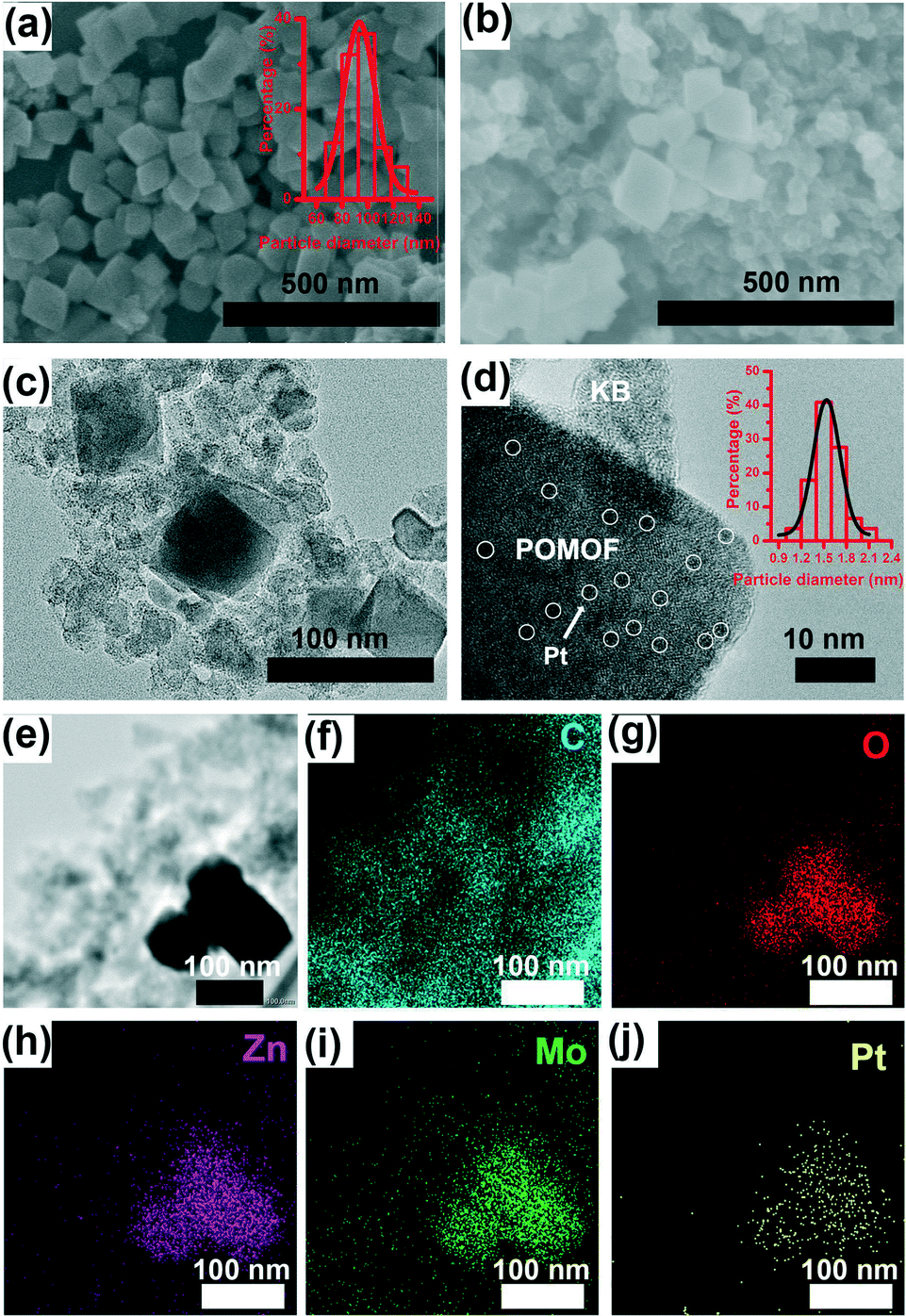 | ||
| Fig. 3 (a) SEM images of POMOF-1 (inset: the particle size distribution of POMOF-1); (b) SEM images of Pt@POMOF-1/KB; (c) TEM images of Pt@POMOF-1/KB; (d) HRTEM images of Pt@POMOF-1/KB; the Pt NPs are highlighted by circles (inset: the particle size distribution of Pt NPs); (e–j) corresponding elemental mapping of C, O, Zn, Mo and Pt of Pt@POMOF-1/KB. | ||
X-Ray powder diffraction patterns (XRD) of POMOF-1 and Pt@POMOF-1/KB are shown in Fig. 4a, in which it is seen that the characteristic peaks of POMOF-1 are retained in the composite, revealing that the loading of Pt and following illumination-reduction process did not affect the crystal structure of POMOF-1. Notably, the XRD peak of Pt metal is not observed, possibly due to the trace amount of Pt NPs in the composite. Moreover, Pt@POMOF-1/KB is also stable in acidic and neutral aqueous solutions at room temperature (Fig. S10†). ICP analysis shows that the Pt content in Pt@POMOF-1/KB is 0.43 wt% (Table S3†).
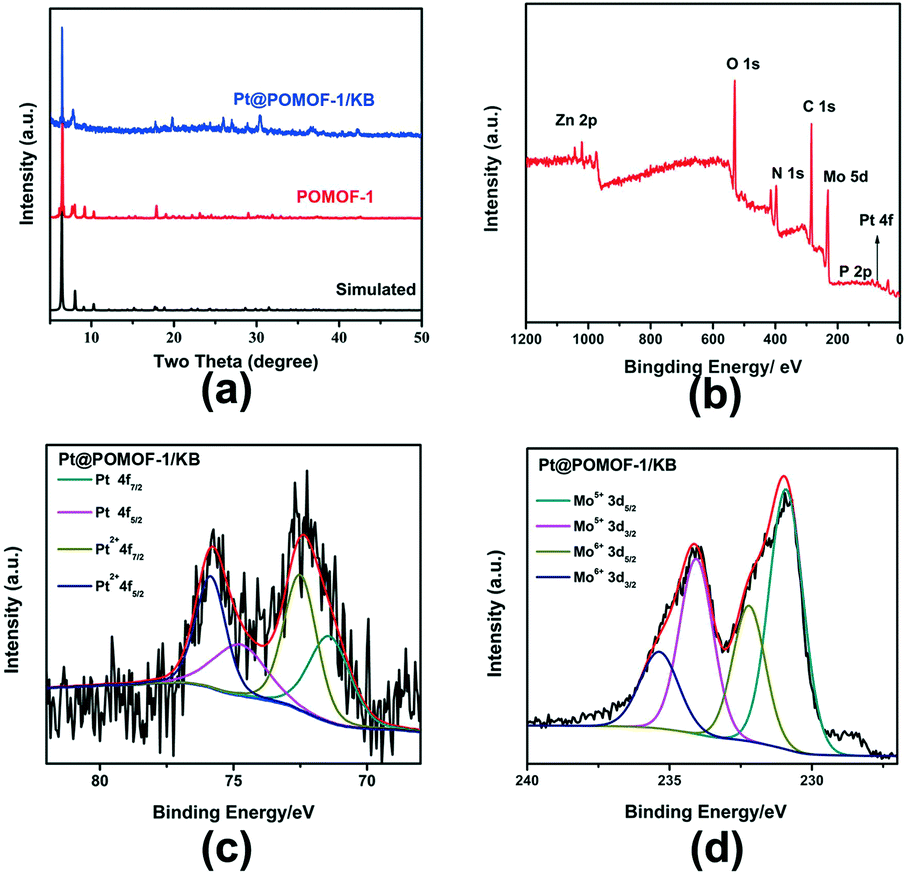 | ||
Fig. 4 (a) Powder X-ray diffraction patterns of POMOF-1 and Pt@POMOF-1/KB; (b) XPS survey spectrum of Pt@POMOF-1/KB; (c) high-resolution XPS spectrum for Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4f; (d) high-resolution XPS spectrum for Mo3d. 4f; (d) high-resolution XPS spectrum for Mo3d. | ||
To further characterize the structure and valence of the Pt@POMOF-1/KB, X-ray photoelectron spectroscopy (XPS) analyses were carried out. Fig. 4b reveals the full XPS survey spectrum of Pt@POMOF-1/KB, which confirms the presence of Zn, Mo, C, N, O, P and Pt elements. The high-resolution XPS spectra for Pt4f can be deconvoluted into four peaks as shown in Fig. 4c. The peaks at 71.3 and 74.6 eV are attributed to Pt4f7/2 and Pt4f5/2 species, respectively. The binding energies at 72.8 and 76.1 eV are assigned to Pt–Ot4f7/2 and Pt–O4f5/2, respectively. The high-resolution XPS spectrum for Mo3d is shown in Fig. 4d. The peaks at 231.2 and 234.4 eV correspond to MoV3d5/2 and MoV3d3/2, respectively. The peaks located at 232.3 and 235.5 eV are assigned to MoVI3d5/2 and MoVI3d3/2, respectively. This result implies that the structure and valence states of POMOF-1 are retained. The above characterization confirms that POMOF-1 has successfully been loaded with Pt NPs. The Brunauer–Emmett–Teller (BET) surface area of Pt@POMOF-1/KB was 826 m2 g−1 (Fig. S11†), which is reduced somewhat relative to KB (1400 m2 g−1). The N2 sorption isotherm exhibits a hysteresis loop, revealing that Pt@POMOF-1/KB has a mesoporous structure, which can expose more active sites, further promoting the HER performance.
2.3 Electrocatalytic HER performance of Pt@POMOF-1/KB
The HER performance of Pt@POMOF-1/KB was evaluated by using a convenient three-electrode system under acidic conditions (0.5 M H2SO4). HER polarization curves of different samples were collected from linear sweep voltammetry (LSV) measurements at a sweep rate of 5 mV s−1. The loading of Pt@POMOF-1/KB and reference samples was 0.849 mg cm−2. As shown in Fig. 5a, the HER activities of POMOF-1, KB and POMOF-1/KB are poor. However, impressively, the Pt@POMOF-1/KB catalyst only requires small overpotentials of 23 and 88 mV to achieve current densities of 10 and 50 mA cm−2, which are comparable to that of 20% Pt/C (50 and 98 mV), and even superior to other reported Pt electrocatalysts (Table S4†) which notably show much higher Pt-loadings of 3–5%, whereas the loading amount of Pt in Pt@POMOF-1/KB is as low as 0.43 wt%. Satisfyingly, the mass activity of Pt@POMOF-1/KB is 100 and 37 times higher than that of commercial 20% Pt/C at overpotentials of 50 and 100 mV (Fig. 5b) as a consequence of the low loading of Pt. This result clearly demonstrates that the good catalytic performance of Pt@POMOF-1/KB can result despite a significantly low noble-metal loading and therefore may meet the cost requirements for large-scale applications. The Faradaic efficiency of Pt@POMOF/KB was measured to be 99% toward HER in 0.5 M H2SO4, showing the favorable HER performance (Fig. S12†).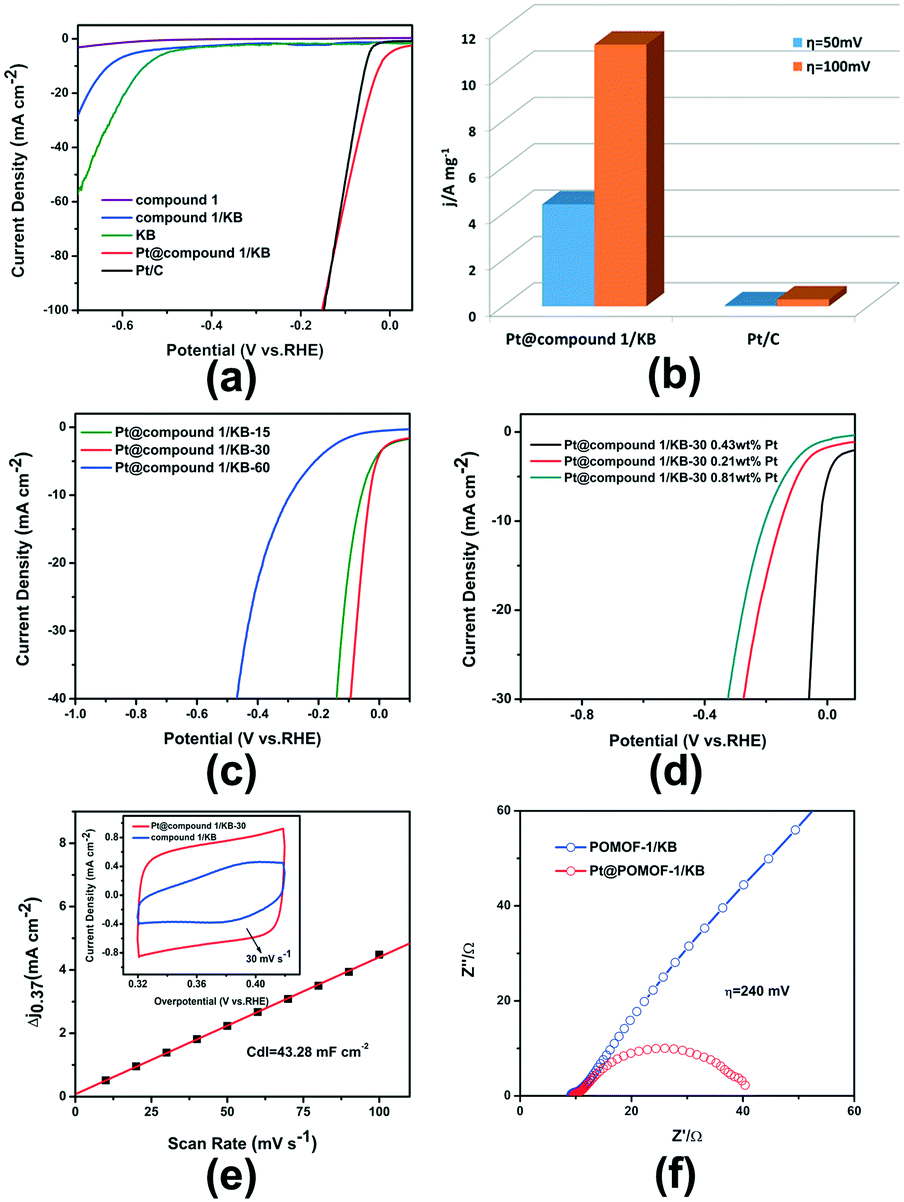 | ||
| Fig. 5 (a) Polarization curves of Pt@POMOF-1/KB, along with POMOF-1, POMOF-1/KB, KB and Pt/C; (b) the mass activity of Pt@POMOF-1/KB and Pt/C; (c) polarization curves of Pt@POMOF-1/KB with different irradiation times (15, 30 and 60 min); (d) polarization curves of Pt@POMOF-1/KB with different Pt loading amounts (0.43, 0.21 and 0.11 wt%); (e) comparison of the CV curves of Pt@POMOF-1/KB-30 and POMOF-1/KB at 30 mV s−1 within the same potential region. Linear fitting of Δj of both samples (Δj = ja − jc) vs. scan rates at a given potential of +0.37 V vs. RHE. ja is the anodic current density and jc is the cathodic current density, respectively. (f) Nyquist plots of Pt@POMOF-1/KB-30 and POMOF-1/KB at an overpotential of 240 mV. | ||
To further understand the superior HER performance of Pt@POMOF-1/KB, we investigate the effect of illumination time on the electrocatalytic performance. Pt@POMOF-1/KB was generally illuminated for 30 min and denoted as Pt@POMOF-1/KB-30. Two other catalysts with different illumination time were also prepared (15 min denoted as Pt@POMOF-1/KB-15 and 60 min denoted as Pt@POMOF-1/KB-60) (Fig. S13–S17†). As indicated in Fig. 5c, when the illumination time is increased to 60 min, the HER activity of Pt@POMOF-1/KB-60 obviously becomes worse and the overpotential for a current density of 10 mA cm−2 increases from 23 to 294 mV, which could be related to the overgrowth of Pt NPs because of the over-reduction of Pt ions (Table S5 and Fig. S17†). Pt@POMOF-1/KB-15 also displays an inferior activity in comparison to Pt@POMOF-1/KB-30. This result may be due to the insufficient reduction of Pt (Table S6 and Fig. S13–S15†), which can be observed from the Pt XPS spectrum. Moreover, an increase of the concentration of Pt from 0.43% to 0.81 wt% (Table S7†) for the precursor also leads to a decrease of HER activity, which stems from the apparent aggregation of Pt NPs (Fig. S18†). A lower concentration of Pt (0.21 wt%, Table S8†) in the precursor also leads to poor HER catalytic activity due to the extremely low amount of Pt loading.
In order to elucidate the origin of the HER ability, the electrochemically active surface area (ECSA) and electrochemical impedance spectroscopy (EIS) were assessed. As depicted in Fig. 5e, Pt@POMOF-1/KB-30 possesses a CV loop with a larger area at a scan rate of 30 mV s−1 than that of POMOF-1/KB, demonstrating that Pt@POMOF-1/KB-30 has a higher double-layer capacitance (Cdl) and a larger active surface area than those of POMOF-1/KB. Furthermore, the electrochemical double-layer capacitance (Cdl) of Pt@POMOF-1/KB-30 was evaluated to be 43.28 mF cm−2, which is also higher than that of Pt@POMOF-1/KB-15 (7.58 mF cm−2) and Pt@POMOF-1/KB-60 (3.40 mF cm−2), implying the high activity of Pt@POMOF-1/KB-30 (Fig. S19†). Electrochemical impedance spectroscopy (EIS) has also been carried out to better understand the catalytic activity. As indicated in Fig. 5f, the Nyquist plot of Pt@POMOF-1/KB-30 exhibits two semicircles, which are fitted to a classical two-time-constant circuit (Fig. S20†). Based on the fitting results, the charge-transfer resistance value (Rct) decreases rapidly as the overpotential increases. Furthermore, the Rct value of Pt@POMOF-1/KB-30 (30 Ω) is much smaller than that of POMOF-1/KB under the same conditions, implying fast electron transport ability of Pt@POMOF-1/KB-30, which promotes the HER performance (Fig. S21†).
To uncover the HER mechanism, the linear portions of the Tafel plots were fitted to the Tafel equation (η = blogj + a, where b is the Tafel slope and j is current density) to acquire the Tafel slopes. As shown in Fig. S22,† the Tafel slope of Pt@POMOF-1/KB-30 was 71.29 mV dec−1, which is lower than that of Pt@POMOF-1/KB-15 (128.43 mV dec−1) and Pt@POMOF-1/KB-60 (195.51 mV dec−1), suggesting fast HER kinetics of Pt@POMOF-1/KB-30. Moreover, on the basis of the Tafel slope range, the hydrogen evolution process occurring on Pt@POMOF-1/KB-30 is via the Volmer–Heyrovsky route, in which the rate-determining step is the release of molecular hydrogen.43
As an important criterion for HER, the long-term durability has been investigated. As shown in Fig. 6, after 1000 cycles continuous cyclic voltammetry sweeps between +0.2 V and −0.2 V, the polarization curve for Pt@POMOF-1/KB-30 exhibits little loss at a current density of 20 mA cm−2. Furthermore, the amperometric i–t curve shown in Fig. 6 also demonstrates that the current density of Pt@POMOF-1/KB-30 displays little decrease after continuous working for 12 h. These results suggest that Pt@POMOF-1/KB-30 possesses good stability in a long-term electrochemical process. Additionally, the XRD pattern and TEM images indicate that the structure of Pt@POMOF-1/KB-30 shows no obvious changes after 1000 cycle sweeps in 0.5 M H2SO4, which may be attributed to the confinement effect of POMOF-1 and interaction between POMOF-1 and Pt NPs (Fig. S23 and S24†).
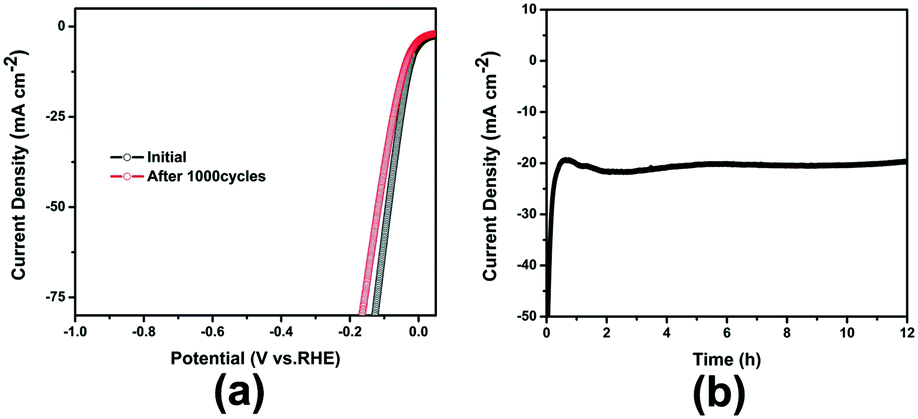 | ||
| Fig. 6 (a) Polarization curves of Pt@POMOF-1/KB-30 initially and after 1000 cycles at 0.5 M H2SO4; (b) time-dependent current density curve of Pt@POMOF-1/KB-30 at overpotential of 50 mV. | ||
Overall, the above measurements confirm the excellent HER performance of Pt@POMOF-1/KB-30 as a low-Pt electrocatalyst. Such remarkable HER performance can be ascribed to the following aspects: (i) the highly reduced POM species in porous POMOF-1 provide an ideal confined space for the adsorption and in situ reduction of Pt ions, resulting in the formation of ultra-small Pt NPs; (ii) the highly reduced POM species endow the whole material with good capability to store H+ and electrons, which may facilitate the transfer of protons and electrons; (iii) the interaction between POM species and Pt NPs enhances the stability of Pt@POMOF-1/KB composite; (iv) the high dispersion of ultra-small Pt nanoparticles enhances the surface area of Pt, which greatly increases the access to active sites of the catalyst.
3 Conclusions
In summary, a new hybrid of low-Pt-loading POMOF and KB carbon (Pt@POMOF-1/KB) have been fabricated. In this hybrid material, the average size of Pt NPs is ca. 1.5 nm and the Pt loading amount is only 0.43 wt%. In 0.5 M H2SO4, Pt@POMOF-1/KB only requires an overpotential of 23 mV to reach a current density of 10 mA cm−2, which is superior to that of 20% Pt/C. The mass activity of Pt@POMOF-1/KB is almost 100 and 37 times more than that of 20% Pt/C at overpotentials of 50 and 100 mV, as a consequence of the low noble-metal loading and therefore may meet the cost requirements for large-scale applications. This work provides a promising approach to minimize the Pt usage and promote its utilization efficiency in HER.4 Experimental section
4.1 Materials and methods
All commercially available reagents for syntheses were used without further purification. H2PtCl6·6H2O was purchased from Shanghai Civi Chemical Technology. H3PO4 was purchased from Beijing Chemical Works. ZnCl2 and (NH4)6Mo7O24·4H2O were purchased from Shanghai Macklin Biochemical. 1,4-Bis(1-imidazolyl)benzene (bimb) was purchased from Jinan Henghua Technology. Mo powder was purchased from Aladdin. Nafion solution (5 wt%) was purchased from Alfa Aesar. The water used throughout all experiments was purified through a Millipore system.4.2 Synthesis of POMOF-1
In a typical preparation of POMOF-1, (NH4)6Mo7O24·4H2O (0.309 g, 0.25 mmol), Mo (0.030 g, 0.31 mmol), H3PO4 (10 μL, 0.147 mmol), ZnCl2 (0.068 g, 0.5 mmol), 25 wt% [Bu4N]OH (200 μL) and bimb ligand (0.010 g, 0.048 mmol) were dissolved in 8 ml deionized water at room temp. under magnetic stirring for 30 min. The solution pH was adjusted to 5–6 with 1 M NaOH. Then the as-prepared solution was transferred to a hydrothermal synthesis reactor and heated at 180 °C for 120 h. The reactor was then cooled to room temperature at a rate of 10 °C h−1 until it reached room temperature. POMOF-1 was obtained as black octahedral crystals (11.2 mg).4.3 Preparation of Pt@POMOF-1/KB-30
In a typical preparation step, 60 mg KB was added into 20 ml ethanol and kept under ultrasonication for 60 min. 20 mg of POMOF-1 was dispersed in 5 ml of 0.1 wt% H2PtCl6 solution. Then the as-prepared mixture was centrifuged at 8000 rpm for 5 min. The obtained black materials were added into isopropanol and stirred strongly under irradiation with a 500 W Xe lamp for 30 min. Then, Pt@POMOF-1 was separated from solution by centrifugation at 8000 rpm and cleaned with deionized water and ethanol for three times. Then Pt@POMOF-1 was added to the above ethanol solution containing 60 mg KB and sonicated for 120 min. The solution was then centrifuged at 9000 rpm for 8 min, and the resulting Pt@POMOF-1/KB composite material was dried in a vacuum drying oven.4.4 Preparation of Pt@POMOF-1/KB-15 and Pt@POMOF-1/KB-60
The preparation method is the same as for Pt@POMOF-1/KB-30 except that the illumination times were 15 and 60 min, respectively.4.5 Preparation of Pt@POMOF-1/KB-0.21 wt% and Pt@POMOF-1/KB-0.81 wt%
The preparation method is same as for Pt@POMOF-1/KB-30 with a Pt content of 0.43 wt% except that the concentrations of H2PtCl6 used were 0.2 and 0.05 wt%, respectively.4.6 Preparation of working electrodes
1 mg Pt@POMOF-1/KB was dispersed in 100 μL of 0.5 wt% Nafion solution. After ultrasonication for 60 min, 6 μL of the homogeneous ink was drop-casted onto a glassy carbon electrode (GCE) with a diameter of 3 mm. The loading of catalyst is about 0.849 mg cm−2. The electrode was dried in air at room temperature.4.7 Electrochemical test
The HER performance of samples was evaluated by a conventional three-electrode system under acidic conditions (0.5 M H2SO4). The glassy carbon electrode (GCE, d = 3 mm) served as a working electrode. A saturated calomel electrode (SCE) was used as the reference electrode, and a graphite rod served as the counter electrode.4.8 X-Ray crystallography
Crystal data and structural refinement for POMOF-1 are listed in Table 1. Selected bond lengths and angles of POMOF-1 are listed in Table S2.†| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. b wR2 = ∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]1/2. | |
|---|---|
| Compound | 1 |
| Formula | C48H40Mo24N16O80P2Zn8 |
| M r | 5008.42 |
| T/K | 293(2) |
| Crystal system | Tetragonal |
| Space group | I41/amd |
| a/Å | 27.560(4) |
| b/Å | 27.560(4) |
| c/Å | 12.012(2) |
| α/° | 90 |
| β/° | 90 |
| γ/° | 90 |
| V/Å3 | 9124(3) |
| Z | 2 |
| μ/mm−1 | 2.700 |
| F(000) | 4716.0 |
| Reflections | 32676 |
| R int | 0.1198 |
| GOF | 1.035 |
| R 1 [I > 2σ(I)] | 0.0669 |
| wR2b (all data) | 0.1782 |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (grant nos. 21671036 and 21771033), Fundamental Research Funds for the Central Universities (grant no. 2412016KJ018), and the Opening Project of Key Laboratory of Polyoxometalate Science of Ministry of Education (grant no. 130028721).Notes and references
- C. Tang, R. Zhang, W. B. Lu, Z. Wang, D. N. Liu, S. Hao, G. Du, A. M. Asiri and X. P. Sun, Angew. Chem., 2017, 129, 860–864 CrossRef
.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. NØrskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed
- Y. Zheng, Y. Jiao, S. Z. Qiao and A. Vasileff, Angew. Chem., Int. Ed., 2018, 57, 7568–7579 CrossRef PubMed
- S. Y. Jing, L. S. Zhang, L. Luo, J. J. Lu, S. B. Yin, P. K. Shen and P. Tsiakarasc, Appl. Catal., B, 2018, 224, 533–540 CrossRef
- Y. Y. Wu, G. D. Li, Y. P. Liu, L. Yang, X. R. Lian, T. Asefa and X. X. Zou, Adv. Funct. Mater., 2016, 26, 4839–4847 CrossRef
- D. Y. Chung, S. W. Jun, G. Yoon, H. Kim, J. M. Yoo, K.-S. Lee, T. Kim, H. Shin, A. K. Sinha, S. G. Kwon, K. Kang, T. Hyeon and Y.-E. Sung, J. Am. Chem. Soc., 2017, 139, 6669–6674 CrossRef PubMed
- Y. F. Cheng, S. K. Lu, F. Liao, L. B. Liu, Y. Q. Li and M. W. Shao, Adv. Funct. Mater., 2017, 27, 1700359 CrossRef
- R. Chen, C. J. Yang, W. Z. Cai, H. Y. Wang, J. W. Miao, L. P. Zhang, S. L. Chen and B. Liu, ACS Energy Lett., 2017, 2, 1070–1075 CrossRef
- M. Ledendecker, H. Schlott, M. Antonietti, B. Meyer and M. Shalom, Adv. Energy Mater., 2017, 7, 1601735 CrossRef
- Y. Y. Chen, Y. Zhang, W. J. Jiang, X. Zhang, Z. H. Dai, L. J. Wan and J. S. Hu, ACS Nano, 2016, 10, 8851–8860 CrossRef PubMed
- K. R. Zhou, J. R. He, X. Q. Wang, J. Lin, Y. Jing, W. L. Zhang and Y. F. Chen, Electrochim. Acta, 2017, 231, 626–631 CrossRef
- S. J. Deng, Y. Zhong, Y. X. Zeng, Y. D. Wang, Z. J. Yao, F. Yang, S. W. Lin, X. L. Wang, X. H. Lu, X. H. Xia and J. P. Tu, Adv. Mater., 2017, 29, 1700748 CrossRef PubMed
- Y. Y. Ma, Z. L. Lang, L. K. Yan, Y. H. Wang, H. Q. Tan, K. Feng, Y. J. Xia, J. Zhong, Y. Liu, Z. H. Kang and Y. G. Li, Energy Environ. Sci., 2018, 11, 2114 RSC
- C. Wu, Y. J. Yang, D. Dong, Y. H. Zhang and J. H. Li, Small, 2017, 13, 1602873 CrossRef PubMed
- J. Q. Yan, H. Wu, P. Li, H. Chen, R. B. Jiang and S. Z. Liu, J. Mater. Chem. A, 2017, 5, 10173–10181 RSC
- P. Nayak, Q. Jiang, N. Kurra, X. B. Wang, U. Buttner and H. N. Alshareef, J. Mater. Chem. A, 2017, 5, 20422–20427 RSC
- M. Tavakkoli, N. Holmberg, R. Kronberg, H. Jiang, J. Sainio, E. I. Kauppinen, T. Kallio and K. Laasonen, ACS Catal., 2017, 7, 3121–3130 CrossRef
- P. T. Wang, X. Zhang, J. Zhang, S. Wan, S. J. Guo, G. Lu, J. I. Yao and X. Q. Huang, Nat. Commun., 2017, 8, 14580 CrossRef PubMed
- J. Wang, Z. Z. Wei, S. J. Mao, H. R. Li and Y. Wang, Energy Environ. Sci., 2018, 11, 800–806 RSC
- J. T. Chen, Y. Yang, J. W. Su, P. Jiang, G. L. Xia and Q. W. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 3596–3601 CrossRef PubMed
- Y. X. Zhang, J. Q. Yan, X. P. Ren, L. Q. Pang, H. Chen and S. Z. Liu, Int. J. Hydrogen Energy, 2017, 42, 5472–5477 CrossRef
- B. B. Jiang, F. Liao, Y. Y. Sun, Y. F. Cheng and M. W. Shao, Nanoscale, 2017, 9, 10138–10144 RSC
- L. S. Xie, X. Ren, Q. Liu, G. W. Cui, R. X. Ge, A. M. Asiri, X. P. Sun, Q. J. Zhang and L. Chen, J. Mater. Chem. A, 2018, 6, 1967–1970 RSC
- L. H. Zhang, L. L. Han, H. X. Liu, X. J. Liu and J. Luo, Angew. Chem., Int. Ed., 2017, 56, 13694–13698 CrossRef PubMed
- Y. Y. Feng, Y. X. Guan, H. J. Zhang, Z. Y. Huang, J. Li, Z. Q. Jiang, X. Gu and Y. Wang, J. Mater. Chem. A, 2018, 6, 11783–11789 RSC
- P. Ghosh, S. K. Saha, A. Roychowdhury and P. Banerjee, Eur. J. Inorg. Chem., 2015, 2851–2857 CrossRef
- P. Ghosh, A. Roychowdhury, M. Corbella, A. Bhaumik, P. Mitra, S. M. Mobin, A. Mukherjee, S. Basuf and P. Banerjee, Dalton Trans., 2014, 43, 13500–13508 RSC
- Y. Zheng, Y. Jiao, Y. H. Zhu, L. H. Li, Y. Han, Y. Chen, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2016, 138, 16174–16181 CrossRef PubMed
- Y. Y. Ma, C. X. Wu, X. J. Feng, H. Q. Tan, L. K. Yan, Y. Liu, Z. H. Kang, E. B. Wang and Y. G. Li, Energy Environ. Sci., 2017, 10, 788–798 RSC
- L. N. Zhang, S. H. Li, H. Q. Tan, S. U. Khan, Y. Y. Ma, H. Y. Zang, Y. H. Wang and Y. G. Li, ACS Appl. Mater. Interfaces, 2017, 9, 16270–16279 CrossRef PubMed
- G. Yan, C. X. Wu, H. Q. Tan, X. J. Feng, L. K. Yan, H. Y. Zang and Y. G. Li, J. Mater. Chem. A, 2017, 5, 765–772 RSC
- Q. Huang, T. Wei, M. Zhang, L. Z. Dong, A. M. Zhang, S. L. Li, W. J. Liu, J. Liu and Y. Q. Lan, J. Mater. Chem. A, 2017, 5, 8477–8483 RSC
- N. Wang, Q. M. Sun, R. S. Bai, X. Li, G. Q. Guo and J. H. Yu, J. Am. Chem. Soc., 2016, 138, 7484–7487 CrossRef PubMed
- T. Yoshida, T. Murayama, N. Sakaguchi, M. Okumura, T. Ishida and M. Haruta, Angew. Chem., 2018, 130, 1539–1543 CrossRef
- H. Medetalibeyoğlu, S. Manap, Ö. A. Yokuş, M. Beytur, F. Kardaş, O. Akyıldırım, V. Özkan, H. Yüksek, M. L. Yola and N. Atar, J. Electrochem. Soc., 2018, 165(5), F338–F341 CrossRef
- P. F. Cao, J. Peng, J. Q. Li and M. L. Zhai, J. Power Sources, 2017, 347, 210–219 CrossRef
- S. Pal, M. Sahoo, V. T. Veettil, K. K. Tadi, A. Ghosh, P. Satyam, R. K. Biroju, P. M. Ajayan, S. K. Nayak and T. N. Narayanan, ACS Catal., 2017, 7, 2676–2684 CrossRef
- S. J. Deng, Y. Zhong, Y. X. Zeng, Y. D. Wang, Z. J. Yao, F. Yang, S. W. Lin, X. Wang, X. H. Lu, X. Xia and J. P. Tu, Adv. Mater., 2017, 29, 1700748 CrossRef PubMed
- D. Z. Wang, J. C. Wang, X. N. Luo, Z. Z. Wu and L. Ye, ACS Sustainable Chem. Eng., 2018, 6, 983–990 CrossRef
-
A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 1998 Search PubMed
- X. J. Yang, M. Sun, H. Y. Zang, Y. Y. Ma, X. J. Feng, H. Q. Tan, Y. H. Wang and Y. G. Li, Chem. – Asian J., 2016, 11, 858–867 CrossRef PubMed
- D. Y. Du, J. S. Qin, T. T. Wang, S. L. Li, Z. M. Su, K. Z. Shao, Y. Q. Lan, X. I. Wang and E. B. Wang, Chem. Sci., 2012, 3, 705–710 RSC
- X. X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details including SEM images, HRTEM images. CCDC 1854604. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ce01151f |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |