
Subsurface nucleation of supercooled acetaminophen†
Limin
Shi
and
Changquan Calvin
Sun
 *
*
Department of Pharmaceutics, University of Minnesota, 308 Harvard St. SE, Minneapolis, MN, USA. E-mail: sunx0053@umn.edu
First published on 5th October 2018
Abstract
The crystallization of supercooled amorphous materials limits their successful applications. The spatial origin of crystallization, nucleation, is one of the most debated topics. We show that homogeneous crystallization may be initiated from tens of micrometers beneath the surface of a supercooled acetaminophen liquid.
Despite crystallization playing a decisive role in a variety of processes of scientific and technological importance, outstanding questions about its nucleation step remain unanswered primarily due to the difficulty in directly observing the nuclei with adequate spatial and temporal resolution.1–3 Numerous simulations as well as theoretical and experimental results posit that crystallization of supercooled liquids and glass materials begins from the surface and propagates inward.4,5 However, other results suggest that the homogeneous crystallization is initiated within supercooled liquids.6–10 For example, the crystallization of a supercooled silicon liquid starts from a few atomic lengths below the surface,6 crystallization of cordierite glass starts near the surface,10 and crystallization occurs in the core of Lennard-Jones systems.7,8 However, evidence concerning the spatial origin of nucleation in organic supercooled liquids is lacking.
Taking advantage of both mechanically-stimulated heterogeneous crystallization11,12 and surface-enhanced crystal growth,13 we have conclusively demonstrated for the first time that homogeneous crystallization is initiated from the subsurface of the supercooled liquid of acetaminophen. A polarized light microscope was used to visualize crystallization. Initially, we prepare supercooled liquids with a thickness of about 1 mm on clean glass slides by cooling the melt of recrystallized acetaminophen crystals at room temperature (24.5 °C), which is about 3 °C above the glass transition temperature of acetaminophen (21.2 ± 0.2 °C). The lack of birefringence confirms the amorphous nature of the fresh sample. A few minutes after the sample is prepared, several small crystals randomly appear in the sample but grow very slowly. We then indent the surface, using a needle, at a point laterally displaced from the sites of previously spontaneously formed crystals. Fast surface crystal growth begins a few seconds after the indentation. While the surface crystal rapidly grows, new crystals spontaneously appear in different regions. The surface crystal growth front sweeps over the sites of the spontaneously formed crystals (Fig. 1a–d, extracted from Movie S1†). The surface crystalline layer contains single crystalline domains as indicated by their uniform color.
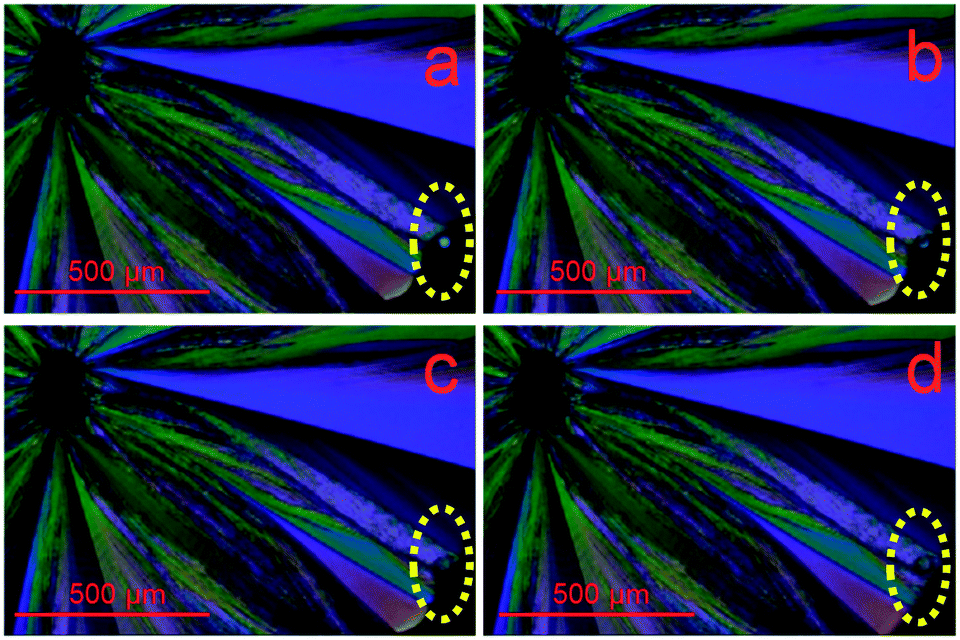 | ||
| Fig. 1 Polarized light microscopy images extracted from a video (Movie S1†) obtained during crystallization of a supercooled acetaminophen liquid. a–d) Fast growing surface crystal sweeps over a spontaneously formed crystal (circled), indicating the subsurface location. | ||
Moreover, the location of the spontaneously formed crystals changes color after the surface crystal layer sweeps over, indicating overlapping between the indentation-induced and spontaneous crystalline regions. These observations can only be explained if the spontaneously formed crystals are located below the surface. If they are formed at the surface, the subsequently grown surface crystal layer will encircle them without causing a color change. In addition, the limited growth of the spontaneously formed crystals is distinct from the rapid and extensive growth of the indentation-induced surface crystal, which is also indicative of their subsurface locations. Using a z-axis calibrated microscope, we determined that the sample stage needed to be moved up by 48.3 ± 7.4 μm (n = 20) to switch focus from the spontaneously formed small crystals to the surface crystal. This suggests that the spontaneous crystallization is started at least that far below the surface under the experimental conditions of 24.5 °C and 42% relative humidity. Subsurface crystals from undisturbed samples eventually emerge and activate the fast surface crystal growth mode. Such surface crystals are symmetrical and radiate outward from the initial crystallization sites (Fig. 2a). At the center of the symmetrical crystal, a broad crater-like feature is observed (Fig. 2b). This is consistent with the subsurface transformation from the supercooled liquid to a higher density crystalline phase. With the growth of the subsurface crystal, an inward tensile stress is generated at the crystal growth front as molecules are rearranged into a denser crystalline phase.14 The build-up of such stress will either form cracks or draw more mobile surface layers inward. The latter will lead to craters as observed here. These craters have a volume of about 1300 μm3. Assuming spherical growth of the subsurface crystal, the distance from the center of the crystal to surface, hn, can be estimated using eqn (1),
 | (1) |
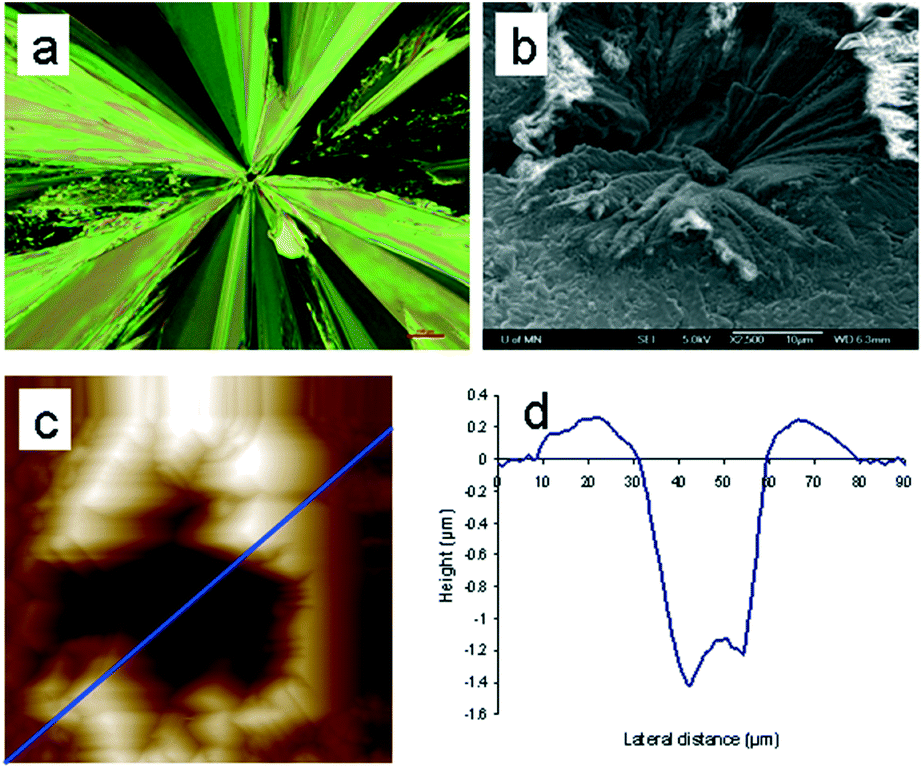 | ||
| Fig. 2 Various microscopy analyses of subsurface crystallization resulting in craters of supercooled acetaminophen liquids in an ambient environment. a, Polarized microscopy image showing the radial growth of the surface crystals from the subsurface nucleation sites. b, SEM image of a crater in the center of a spontaneous crystallization site. An island can be seen at the center of the crater. The formation of the crater is a result of both the subsurface crystallization and density difference between the supercooled liquid and crystalline solid. c, AFM topographical image (70 μm × 70 μm) of a crystal crater. Large material pileup at the perimeter of the crater is also observed. d, Cross-sectional profile of the crystal crater, along the blue line in image c, showing that both the pileup and island can be as high as several hundreds of nanometers above the reference surfaces. | ||
This interesting phenomenon cannot be satisfactorily explained by the prevailing nucleation mechanisms.4,5,12,14–18 First, the homogeneous subsurface crystallization of pure supercooled acetaminophen liquids is far from the substrate. This makes the mechanism of heterogeneous crystallization due to the substrate unlikely.15,18 Second, because we observe this phenomenon in samples free from external stresses, the mechanisms of molecule reordering,16 flow dilatation,12 and large local temperature rise induced by external stresses17 are also unlikely. Third, the enrichment of one component in a mixture, i.e., the surface layering mechanism, is not applicable to pure supercooled organic liquid systems.4,5 Lastly, the ‘elastic-strain’ mechanism does not play any significant role because, at a temperature above Tg, supercooled liquids relieve any residual strain readily.14
To gain more insight into the subsurface nucleation phenomenon, we have considered both thermodynamic and kinetic factors. In the classic view, nucleation results in a net free energy change of the system involving energy loss by forming a crystalline phase from the supercooled liquid and energy gain by creating a new crystal–liquid interface.19 The critical nucleus size, r*, may be calculated using eqn (2),3
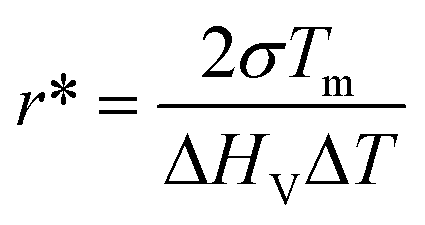 | (2) |
The crystal–liquid interfacial energy, σ, can be estimated using Turnbull's equation (eqn (3)),20
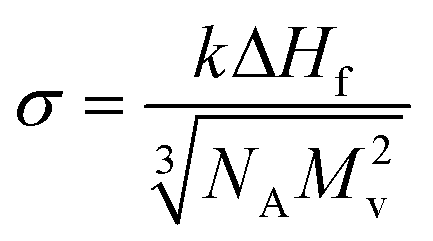 | (3) |
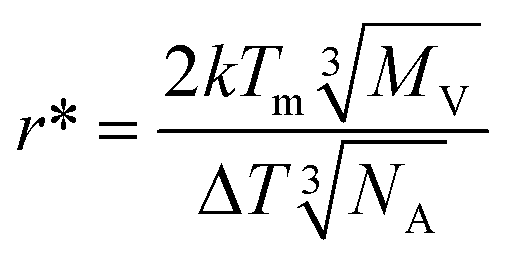 | (4) |
It is known that the molecular mobility at the surface is much higher than that in the bulk of supercooled organic liquids at a given temperature.13,21–24 In other words, surface molecules exhibit mobility similar to bulk molecules at a higher temperature. It is reasonable to expect that the molecular mobility progressively decreases when moving from the surface to bulk, i.e., the temperature corresponding to molecular mobility decreases from the surface to bulk. Hence, the effective degree of supercooling increases from the surface to bulk. For the supercooled acetaminophen liquid at 24.5 °C, the measured crystal growth rate, μ, at the bulk (measured using a sample sandwiched between a glass slide and a cover glass), subsurface, and surface is 0.55 ± 0.03, 140.9 ± 8.8, and 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 488 ± 643 nm s−1, respectively. Based on the reported bulk crystal growth rate profile of supercooled acetaminophen liquids,25 the observed surface and subsurface crystal growth rates correspond to bulk growth rates at 86 and 62 °C, respectively. The corresponding degrees of supercooling at the surface, subsurface, and bulk are 82, 106, and 144 °C. These lead to r* values of 1.99, 1.54, and 1.13 nm, respectively. Therefore, bulk is the thermodynamically preferred nucleation site. However, nucleation is also affected by the kinetic factor molecular mobility. Higher mobility increases the probability of the molecules to form a cluster with a size beyond r*. Since the molecular mobility in the bulk of a supercooled liquid is much lower than that on the surface, nucleation deep in the sample is kinetically disfavored. Fig. 3 illustrates these opposing effects that naturally lead to an optimum nucleation location below the surface. This behavior is analogous to the classical bell-shaped nucleation rate distribution below the melting point.26 This implies that improved stability of amorphous solids can be obtained if the material is thinner than the depth of spontaneous nucleation at the corresponding temperature. Since the interplay between the kinetics and thermodynamics of the process is a general phenomenon, the subsurface nucleation may be broadly relevant to other supercooled liquids. Further understanding of this phenomenon may lead to new strategies to more effectively stabilize amorphous materials.
488 ± 643 nm s−1, respectively. Based on the reported bulk crystal growth rate profile of supercooled acetaminophen liquids,25 the observed surface and subsurface crystal growth rates correspond to bulk growth rates at 86 and 62 °C, respectively. The corresponding degrees of supercooling at the surface, subsurface, and bulk are 82, 106, and 144 °C. These lead to r* values of 1.99, 1.54, and 1.13 nm, respectively. Therefore, bulk is the thermodynamically preferred nucleation site. However, nucleation is also affected by the kinetic factor molecular mobility. Higher mobility increases the probability of the molecules to form a cluster with a size beyond r*. Since the molecular mobility in the bulk of a supercooled liquid is much lower than that on the surface, nucleation deep in the sample is kinetically disfavored. Fig. 3 illustrates these opposing effects that naturally lead to an optimum nucleation location below the surface. This behavior is analogous to the classical bell-shaped nucleation rate distribution below the melting point.26 This implies that improved stability of amorphous solids can be obtained if the material is thinner than the depth of spontaneous nucleation at the corresponding temperature. Since the interplay between the kinetics and thermodynamics of the process is a general phenomenon, the subsurface nucleation may be broadly relevant to other supercooled liquids. Further understanding of this phenomenon may lead to new strategies to more effectively stabilize amorphous materials.
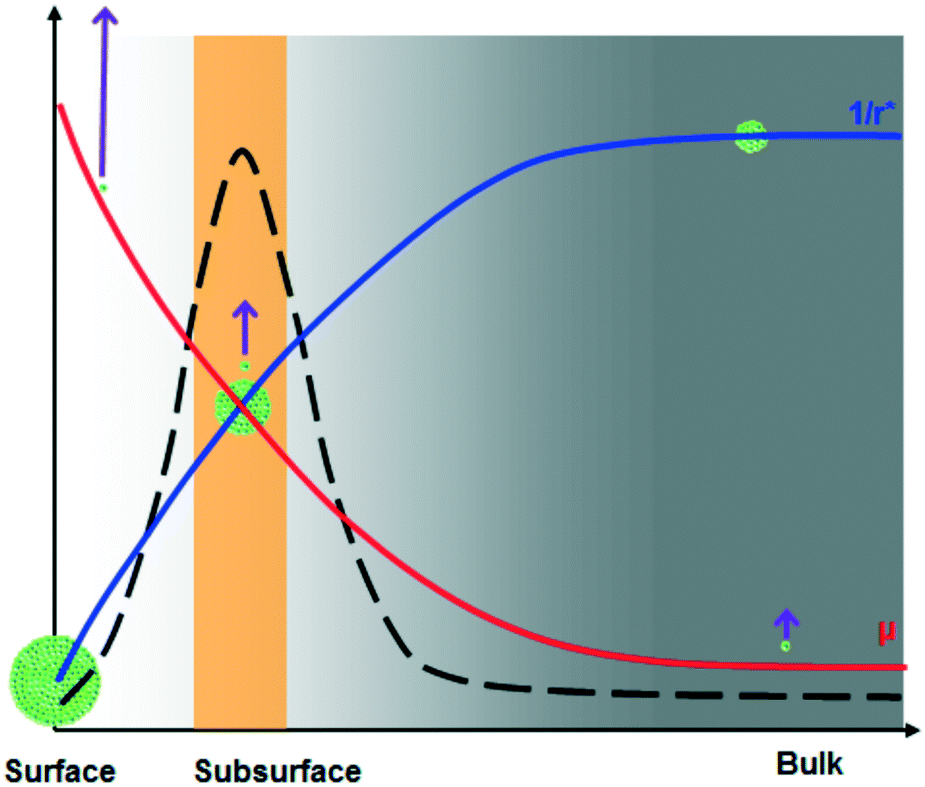 | ||
| Fig. 3 Schematic illustration of the distribution of molecular mobility (μ) and reciprocal critical nuclei size (1/r*) with depth in supercooled liquids and glass materials. The large critical nucleus size (green clusters) limits nucleation on the surface, while the slower molecular mobility (upward arrows) hinders nucleation in the bulk. The optimum nucleation site lies in the subsurface region as a result of the interplay between these kinetic and thermodynamic factors. The broken line suggests the possible depth-dependent nucleation rate distribution. | ||
Conflicts of interest
There are no conflicts to declare.References
- P. G. Vekilov, Nat. Mater., 2012, 11, 838–840 CrossRef CAS PubMed
.
- F. Schüth, P. Bussian, P. Ågren, S. Schunk and M. Lindén, Solid State Sci., 2001, 3, 801–808 CrossRef
-
J. W. P. Schmelzer, Nucleation theory and applications, Wiley-VCH, 2005 Search PubMed
- P. W. Sutter and E. A. Sutter, Nat. Mater., 2007, 6, 363–366 CrossRef CAS PubMed
- O. G. Shpyrko, R. Streitel, V. S. K. Balagurusamy, A. Y. Grigoriev, M. Deutsch, B. M. Ocko, M. Meron, B. Lin and P. S. Pershan, Science, 2006, 313, 77–80 CrossRef CAS PubMed
- T. Li, D. Donadio, L. M. Ghiringhelli and G. Galli, Nat. Mater., 2009, 8, 726–730 CrossRef CAS PubMed
- P. R. t. Wolde and D. Frenkel, Science, 1997, 277, 1975–1978 CrossRef PubMed
- D. Erdemir, A. Y. Lee and A. S. Myerson, Acc. Chem. Res., 2009, 42, 621–629 CrossRef CAS PubMed
- P. G. Vekilov, Cryst. Growth Des., 2004, 4, 671–685 CrossRef CAS
- I. Avramov and G. Voelksch, J. Non-Cryst. Solids, 2002, 304, 25–30 CrossRef CAS
- J. Sort, L. F. Bonavina, A. Varea, C. Souza, W. J. Botta, C. S. Kiminami, C. Bolfarini, S. Suriñach, M. D. Baró and J. Nogués, Small, 2010, 6, 1543–1549 CrossRef CAS PubMed
- J. J. Kim, Y. Choi, S. Suresh and A. S. Argon, Science, 2002, 295, 654–657 CAS
- T. Wu and L. Yu, Pharm. Res., 2006, 23, 2350–2355 CrossRef CAS PubMed
- J. Schmelzer, R. Pascova, J. Möller and I. Gutzow, J. Non-Cryst. Solids, 1993, 162, 26–39 CrossRef CAS
- V. Halka, R. Streitel and W. Freyland, J. Phys.: Condens. Matter, 2008, 20, 355007 CrossRef
- H. Chen, Y. He, G. J. Shiflet and S. J. Poon, Nature, 1994, 367, 541–543 CrossRef CAS
- H. J. Leamy, T. T. Wang and H. S. Chen, Metall. Trans., 1972, 3, 699–708 CrossRef CAS
- M. V. Akdeniz and A. O. Mekhrabov, J. Nanosci. Nanotechnol., 2008, 8, 894–900 CrossRef CAS PubMed
-
P. G. Debenedetti, Metastable Liquids: concepts and principles, Princeton University Press, 1996 Search PubMed
- D. Turnbull, J. Appl. Phys., 1950, 21, 1022–1028 CrossRef CAS
- Z. Fakhraai and J. A. Forrest, Science, 2008, 319, 600–604 CrossRef CAS PubMed
- Y. Sun, L. Zhu, K. L. Kearns, M. D. Ediger and L. Yu, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5990–5995 CrossRef CAS PubMed
- R. D. Priestley, C. J. Ellison, L. J. Broadbelt and J. M. Torkelson, Science, 2005, 309, 456–459 CrossRef CAS PubMed
- L. Zhu, C. W. Brian, S. F. Swallen, P. T. Straus, M. D. Ediger and L. Yu, Phys. Rev. Lett., 2011, 106, 256103 CrossRef CAS PubMed
- N. S. Trasi and L. S. Taylor, CrystEngComm, 2012, 14, 5188–5197 RSC
- N. Okamoto and M. Oguni, Solid State Commun., 1996, 99, 53–56 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, a movie clip showing fast surface crystal growth sweeping over subsurface nuclei, and the calibration curve for the Z-axis of the microscope. See DOI: 10.1039/c8ce01148f |
| This journal is © The Royal Society of Chemistry 2018 |