
Thermal expansion along one-dimensional chains and two-dimensional sheets within co-crystals based on halogen or hydrogen bonds†
Kristin M.
Hutchins
 *a,
Daniel K.
Unruh
a,
Dontrell D.
Carpenter
b and
Ryan H.
Groeneman
*b
*a,
Daniel K.
Unruh
a,
Dontrell D.
Carpenter
b and
Ryan H.
Groeneman
*b
aDepartment of Chemistry and Biochemistry, Texas Tech University, Lubbock, TX 79409, USA. E-mail: kristin.hutchins@ttu.edu; Tel: (+1) 806 834 2744
bDepartment of Biological Sciences, Webster University, St. Louis, MO 63119, USA. E-mail: ryangroeneman19@webster.edu; Tel: (+1) 314 246 7466
First published on 30th August 2018
Abstract
The ability to vary the thermal expansion parameters within halogen- or hydrogen-bonded co-crystals is reported. An acceptor molecule, 1,2-bis(4-pyridyl)acetylene, was selected due to its ability to form supramolecular one-dimensional chains, regardless of the ditopic donor type. We demonstrate that the smallest expansion occurs along the chain, and greater expansion occurs within the two-dimensional sheet, which is based on weaker halogen interactions.
The ability to control the physical and chemical properties of molecular solids continues to be an important area of research within materials science.1 Co-crystallization strategies, preparation of new solid phases through the combination of two or more neutral solids, have proven to be a reliable approach to modify the properties of these materials.2 These properties are generally different compared to those of either of the single-component solids. The introduction of a second component gives rise to different crystal packing arrangements, and the presence of additional non-covalent interactions will ultimately influence the resulting properties of the new material.
To this end, it has been previously reported that small modifications to the hydrogen- or halogen-bond donor or acceptor molecules can alter the thermal expansion (TE) properties of various co-crystals. For example, Metrangolo and Resnati reported co-crystals based on trans-1,2-bis(4-pyridyl)ethylene (4,4′-BPE) with two different halogen-bond donors, namely, 1,4-diiodoperfluorobenzene (C6I2F4) and 1,4-dibromoperfluorobenzene (C6Br2F4), that yielded isostructural, one-dimensional (1D) chains.3 The TE was calculated using the observed changes in bond lengths as a function of temperature where the I⋯N halogen bond was shown to be stronger than the Br⋯N bond in these solids. Recently, TE properties were varied by changing the strength of halogen-bonding interactions within a series of co-crystals based on 4,6-diX-resorcinol (where: X = Cl, Br, I) and 4,4′-azopyridine.4 By changing the halogen atoms on the resorcinol, the type and strength of the halogen bond was tuned and, in turn, the TE parameters. Modification of hydrogen-bond strength has been investigated within 1D chains based on 1,2,3,4-cyclobutanetetracarboxylic acid with 4,4′-BPE, wherein Saha reported that the stronger O–H⋯N hydrogen bond undergoes less TE than the weaker C–H⋯O interactions in the solid.5
Here, we report the ability to influence TE by varying the type of non-covalent interaction that assembles donor and acceptor molecules into 1D chains (Scheme 1). The chains are held together by either halogen or hydrogen bonds between the donor molecule and 1,2-bis(4-pyridyl)acetylene (4,4′-BPA) as the acceptor molecule. The focus of this study was to determine how the TE would vary based on the type and strength of the non-covalent interaction. We selected the halogen-bond donors C6I2F4 and C6Br2F4 due to their ability to form reliable interactions with various pyridines.6 The hydrogen-bond donor 1,2,4,5-tetrabromobenzene (C6Br4H2) was chosen because of its reported ability to form a hydrogen-bonded 1D chain with 4,4′-BPE, a similarly shaped bipyridine.7
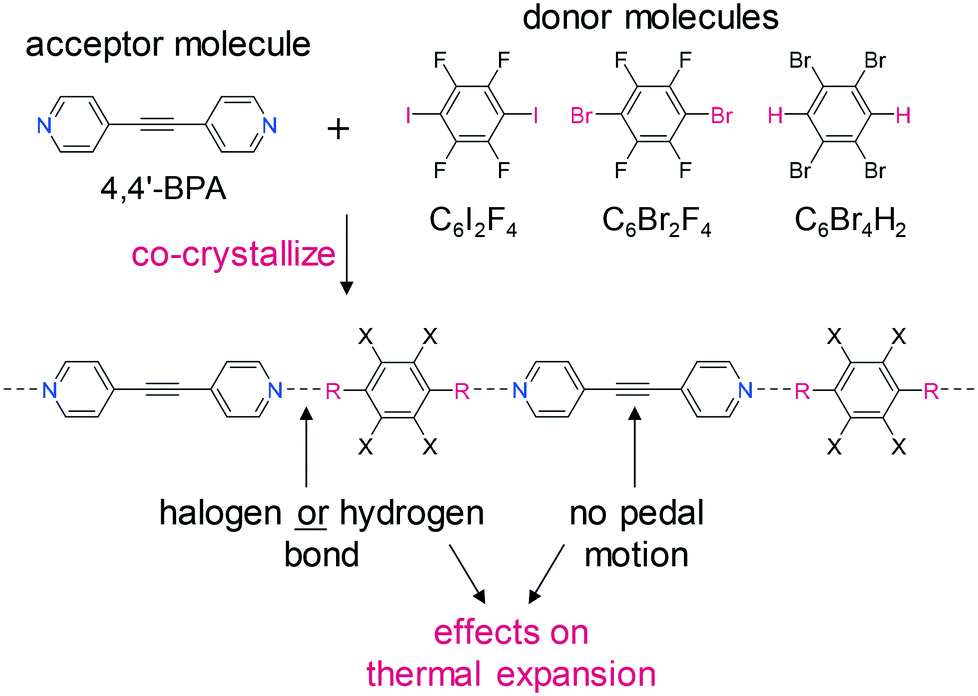 | ||
| Scheme 1 Components of the co-crystals and the 1D chain that form via the co-crystallization process. | ||
We have recently demonstrated that the molecular pedal motion of olefin and azo groups contributes to TE in co-crystals.8 Thus, to remove the effects of pedal motion from the TE parameters and focus primarily on non-covalent interactions, an acetylene-containing molecule, 4,4′-BPA, was selected as the acceptor molecule because it cannot undergo pedal motion (Scheme 1). In addition to the 1D halogen- or hydrogen-bonded chains, the components of the co-crystals are further assembled into two-dimensional (2D) sheets via halogen interactions involving the substituents on the benzene ring. The principal axis (i.e. strongest interaction) for the co-crystals sustained by 1D halogen bonds is observed along the vector of the 1D polymeric chains, and the principal axis for the co-crystal assembled by 1D hydrogen bonds lies along the 1D polymeric chains. More expansion occurs within the 2D sheet, which is based on weaker halogen interactions.
Co-crystallization was achieved by dissolving 25 mg of 4,4′-BPA in 2 mL of toluene and combining the solution with a separate solution containing the appropriate donor (C6I2F4, C6Br2F4, or C6Br4H2; 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio), which was also dissolved in 2 mL of toluene. The combined solutions were allowed to evaporate slowly, and crystals suitable for X-ray diffraction‡ formed within two days for each co-crystal.
:1 molar ratio), which was also dissolved in 2 mL of toluene. The combined solutions were allowed to evaporate slowly, and crystals suitable for X-ray diffraction‡ formed within two days for each co-crystal.
Single-crystal X-ray analyses determined the components of each co-crystal to be present in a 1:1 ratio, namely, (C6I2F4)·(4,4′-BPA), (C6Br2F4)·(4,4′-BPA), and (C6Br4H2)·(4,4′-BPA). All three co-crystals crystallize in different centrosymmetric space groups.
In the case of (C6I2F4)·(4,4′-BPA), the solid crystallizes in the orthorhombic space group Pnnm, and one-fourth of each molecule is found in the asymmetric unit. X-ray diffraction data revealed that the two components of the co-crystal assembled into a 1D chain held together by I⋯N halogen bonds [I⋯N (Å): 2.586(2) at 270 K] (Fig. 1a). Within the chain, the pyridine and perfluorobenzene rings are found to be nearly co-planar, only deviating from the planarity by 0.85°. The chains stack slightly offset, such that the iodine atoms lie between a pyridine and perfluorobenzene ring (Fig. 1a). The offset stacking may have arisen from the negative electrostatic potential region of the halogen interacting with the electron deficient pyridine ring. The stacked chains interact with neighbouring stacks that lie nearly perpendicular (89.92°) via C–H⋯F forces [C⋯F (Å): 3.170(2), 3.264(2) at 270 K] (Fig. 1b). All fluorine atoms engage in this interaction to produce a 2D sheet, with the C–H⋯F forces extending along the crystallographic c-axis.
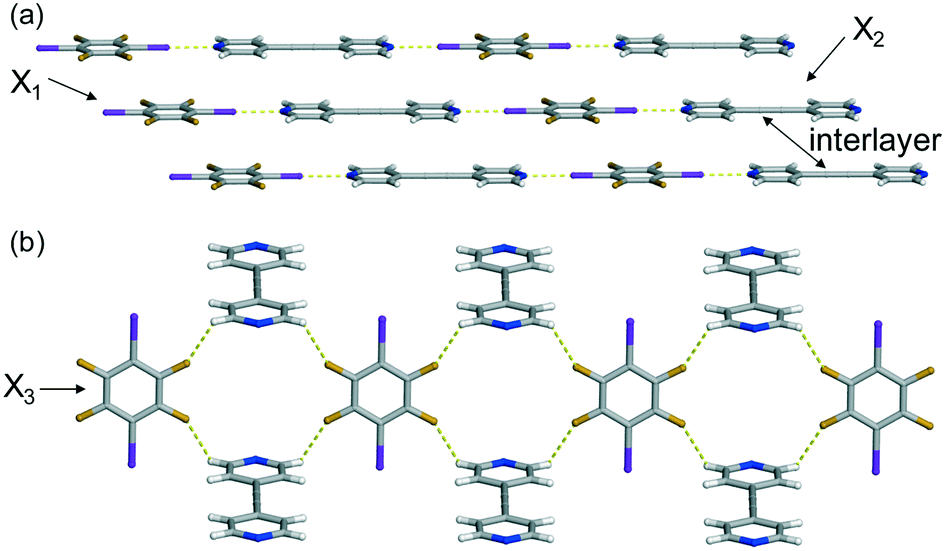 | ||
| Fig. 1 X-ray crystal structure of (C6I2F4)·(4,4′-BPA) illustrating: (a) the I⋯N halogen bonds and (b) the C–H⋯F forces that produce the 2D sheet. Thermal expansion axes (X1–X3) are denoted. | ||
The co-crystal (C6Br2F4)·(4,4′-BPA) crystallizes in the monoclinic space group P21/c and the asymmetric unit contains half of each molecule. Again, the single-crystal X-ray diffraction data revealed that the co-crystal is also a 1D polymer, but this time the chains are held together by Br⋯N halogen bonds [Br⋯N (Å): 2.893(2) at 270 K] (Fig. 2a). Unlike the previous halogen-bonded polymer, the aromatic rings along the chain of (C6Br2F4)·(4,4′-BPA) deviate from the planarity by 18.58°. The chains stack in a similar fashion, and, similarly, the stacked bromine atoms are neighboured by a pyridine and perfluorobenzene ring. The perfluorobenzene interacts with two 4,4′-BPA molecules via C–H⋯F forces [C⋯F (Å): 3.401(3) at 270 K] resulting in a 2D sheet-like structure that extends along the crystallographic b-axis (Fig. 2b). These perfluorobenzene and pyridine rings are nearly perpendicular with a measured angle of 87.26°.
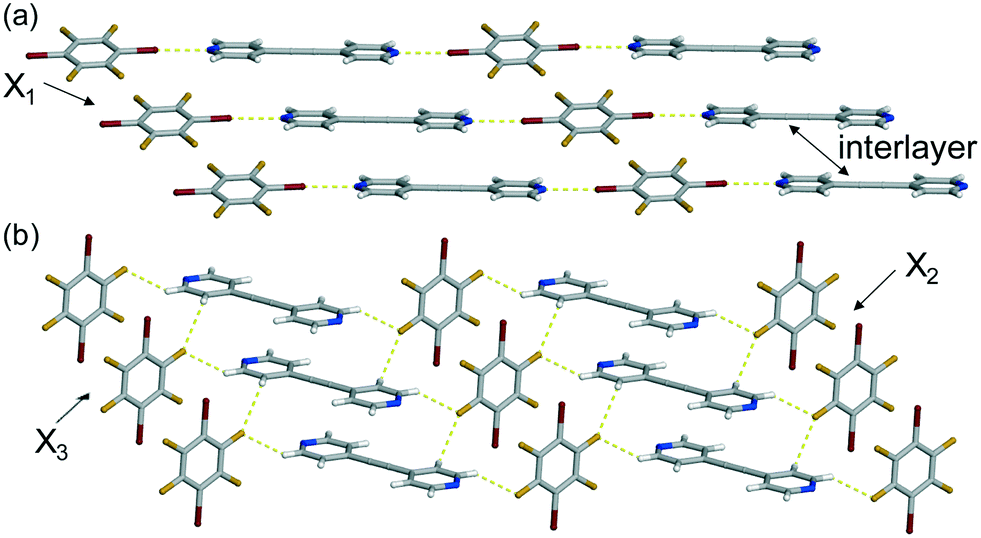 | ||
| Fig. 2 X-ray crystal structure of (C6Br2F4)·(4,4′-BPA) illustrating: (a) the Br⋯N halogen bonds and (b) the C–H⋯F forces that produce the 2D sheet. Thermal expansion axes (X1–X3) are denoted. | ||
The final co-crystal, (C6Br4H2)·(4,4′-BPA), crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) .§ The asymmetric unit contains one-half of two unique C6Br4H2 molecules and one-half of two unique 4,4′-BPA molecules. Single-crystal X-ray diffraction data revealed that the co-crystal is also a 1D polymer sustained by C–H⋯N hydrogen bonds [C⋯N (Å): 3.345(4) and 3.394(4) at 270 K] (Fig. 3a). The chains exhibit a slight zigzag pattern running along the hydrogen-bond axis. Both the C6Br4H2 and 4,4′-BPA molecules are found to engage in homogeneous face-to-face π–π stacking along the crystallographic a-axis, as seen in similar co-crystals based on this donor (Fig. 3a).7 Turning to the extended structure, the bromine atoms on neighbouring C6Br4H2 molecules interact via type I Br⋯Br contacts9 [Br⋯Br (Å): 3.678(1), θ1 = 156.96°, θ2 = 141.13° at 270 K] (Fig. 3b) to generate a 2D sheet. As a result of these Br⋯Br contacts, the nearest neighbouring C6Br4H2 molecules are arranged in a stair-like pattern where the phenyl rings are twisted by 41.83°.
.§ The asymmetric unit contains one-half of two unique C6Br4H2 molecules and one-half of two unique 4,4′-BPA molecules. Single-crystal X-ray diffraction data revealed that the co-crystal is also a 1D polymer sustained by C–H⋯N hydrogen bonds [C⋯N (Å): 3.345(4) and 3.394(4) at 270 K] (Fig. 3a). The chains exhibit a slight zigzag pattern running along the hydrogen-bond axis. Both the C6Br4H2 and 4,4′-BPA molecules are found to engage in homogeneous face-to-face π–π stacking along the crystallographic a-axis, as seen in similar co-crystals based on this donor (Fig. 3a).7 Turning to the extended structure, the bromine atoms on neighbouring C6Br4H2 molecules interact via type I Br⋯Br contacts9 [Br⋯Br (Å): 3.678(1), θ1 = 156.96°, θ2 = 141.13° at 270 K] (Fig. 3b) to generate a 2D sheet. As a result of these Br⋯Br contacts, the nearest neighbouring C6Br4H2 molecules are arranged in a stair-like pattern where the phenyl rings are twisted by 41.83°.
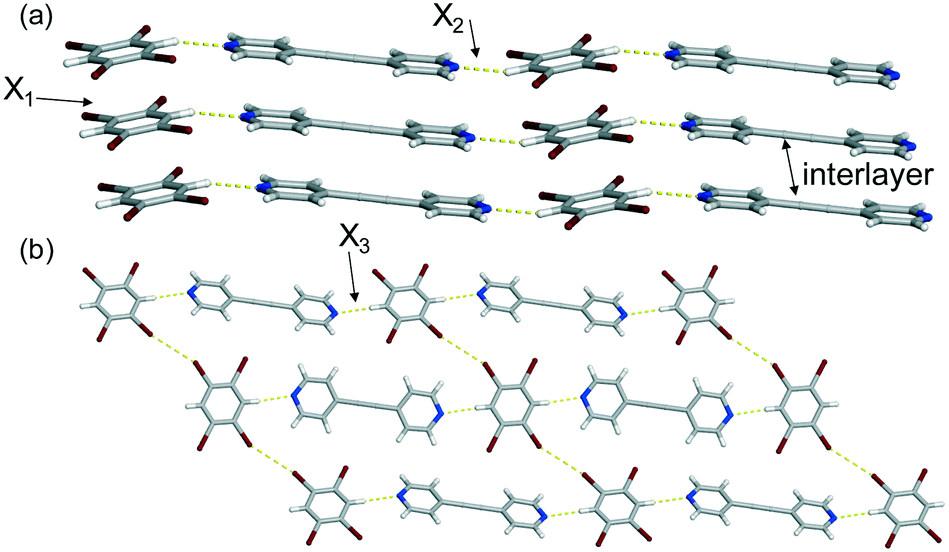 | ||
| Fig. 3 X-ray crystal structure of (C6Br4H2)·(4,4′-BPA) illustrating (a) the C–H⋯N hydrogen bonds and (b) the Br⋯Br contacts that produce the 2D sheet. Thermal expansion axes (X1–X3) are denoted. | ||
In order to determine the TE values, a variable temperature crystallographic study was performed on the same crystal with five data sets collected at: 270, 250, 230, 210, and 190 K. The first data set in each case was collected at 190 K, and then the crystal was warmed to the additional temperatures. PASCal10 was employed to calculate the TE coefficients and the principal axes of each co-crystal over the temperature range of 270 to 190 K. If the crystallographic axis length increases upon heating, positive TE has occurred, while a decrease in the axis length upon heating corresponds to negative TE.11 It is well-known that stronger interactions are less affected by temperature changes, and weaker interactions are more affected.11
The least TE (X1 axis) occurs along the crystallographic b-axis for (C6I2F4)·(4,4′-BPA) and (C6Br2F4)·(4,4′-BPA) and primarily the c-axis for (C6Br4H2)·(4,4′-BPA) (Table 1). The positive and negative TE values correspond to the changes in the distance between the layers of the 1D chains upon heating. The co-crystal (C6I2F4)·(4,4′-BPA) undergoes a minimal increase (0.001 Å) in the distance between layers, corresponding to a very small positive TE value (Fig. 1a). On the other hand, the co-crystals (C6Br2F4)·(4,4′-BPA) and (C6Br4H2)·(4,4′-BPA) undergo small decreases in the distance between the layers of the 1D chains upon heating. Although the decrease is small, the overall length change in (C6Br2F4)·(4,4′-BPA) (0.009 Å) is approximately double that in (C6Br4H2)·(4,4′-BPA) (0.004 Å), which is in agreement with the TE values (Fig. 2a and 3a).
| Co-crystal | α X 1 (MK−1) [axis] | α X 2 (MK−1) [axis] | α X 3 (MK−1) [axis] | α V (MK−1) |
|---|---|---|---|---|
| (C6I2F4)·(4,4′-BPA) | 4(1) | 46(3) | 124(4) | 177 |
| [0 1 0] | [−1 0 0] | [0 0 1] | ||
| (C6Br2F4)·(4,4′-BPA) | −25(4) | 58(2) | 121(1) | 155 |
| [0 1 0] | [−3 0 –1] | [1 0 5] | ||
| (C6Br4H2)·(4,4′-BPA) | −13(1) | 69(1) | 143(2) | 200 |
| [0 1 2] | [2 2 −1] | [−5 1 0] |
The supramolecular interactions that contribute to the X1 axis include primarily either the halogen or hydrogen bonds that produce the 1D supramolecular chain. The halogen or hydrogen bonding interaction is the strongest non-covalent force within each co-crystal; thus, these bonds experience the smallest change in length when the temperature is altered. Upon heating, the strongest N⋯I halogen bonds underwent the smallest increase in length (0.015 Å), followed by the C–H⋯N hydrogen bonds (avg. 0.018 Å), and the N⋯Br halogen bonds experienced the largest change (0.029 Å).
In all three co-crystals, the X2 axis encompasses primarily the supramolecular interactions that constitute the 2D sheets. In the case of (C6I2F4)·(4,4′-BPA) and (C6Br2F4)·(4,4′-BPA), the interactions include halogen bonds, C–H⋯F, and weak C(benzene or pyridine)⋯X (between stacked chains) forces. The halogen bonds and C–H⋯F forces are stronger in (C6I2F4)·(4,4′-BPA) (see ESI,† Table S8). All the interaction lengths in both co-crystals increased upon heating; however, the changes were smaller for the stronger forces in (C6I2F4)·(4,4′-BPA) (ca. 0.01–0.04 Å) when compared to those in (C6Br2F4)·(4,4′-BPA) (ca. 0.02–0.04 Å). Thus, (C6Br2F4)·(4,4′-BPA) exhibits a larger TE coefficient. In the hydrogen-bonded co-crystal (C6Br4H2)·(4,4′-BPA), the contributing interactions involve weaker C–H⋯Br, Br⋯Br, and π⋯π forces. All these interactions were more affected by heating and their lengths increased by ca. 0.03–0.05 Å, which is supportive of a slightly larger TE coefficient compared to that of the halogen-bonded co-crystals.
Most expansion occurs along the X3 axis, which is composed of the weakest interactions sustaining the co-crystals. In the case of (C6I2F4)·(4,4′-BPA), the C–H⋯F interactions contribute primarily to the expansion and their lengths increase upon heating by ca. 0.04 Å. For the co-crystal (C6Br2F4)·(4,4′-BPA), the C(benzene)⋯F and C(benzene or pyridine)⋯Br (between stacked chains) forces lie along the X3 axis and lengthen upon heating by ca. 0.03 Å and 0.02 Å, respectively. The N⋯Br halogen bonds also contribute slightly to the X3 axis, lengthening upon heating by ca. 0.03 Å. The overall similar changes in these interaction distances results in similar TE values for the two halogen-bonded co-crystals. The TE value along X3 for the hydrogen-bonded co-crystal is larger, however. The interactions that contribute to the TE in (C6Br4H2)·(4,4′-BPA) are C–H⋯Br, C(benzene)⋯Br, and π⋯π, all increasing in length upon heating by ca. 0.03, 0.05, and 0.05 Å, respectively.
Using strong, directional halogen or hydrogen bonds to assemble components in co-crystals can provide a means to control TE along the bonding directions, here, the 1D chains and 2D sheets (X1 and X2 axes). Minimal TE occurs along the strongest, non-covalent interaction bonding directions, while TE occurs to a larger degree along the weaker interactions that sustain the co-crystal. In the co-crystals reported here, we have achieved control over two dimensions; however, the ability to design and realize full control over the intermolecular interactions that sustain co-crystals in all three dimensions will aid in achieving predictable TE properties.
Conclusions
In this report, we describe the TE parameters of co-crystals assembled into 1D chains via either halogen or hydrogen bonds. In all cases, the least expansion occurs along the halogen- or hydrogen-bonded chain. The 2D sheet structure in each co-crystal is sustained by weaker interactions, and more expansion occurs within the sheet structure. It is well-known that both halogen and hydrogen bonds can be utilized to modify TE properties; however, only a few studies compare them as in this report. We are currently expanding this study to 2D systems that are held together by stronger halogen- or hydrogen-bonding interactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. M. H gratefully acknowledges financial support from Texas Tech University in the form of start-up funding. R. H. G. gratefully acknowledges financial support from Webster University in the form of various Faculty Research Grants.Notes and references
-
(a) M. I. Tamboli, D. P. Karothu, M. S. Shashidhar, R. G. Gonnade and P. Naumov, Chem. – Eur. J., 2018, 24, 4133 CrossRef PubMed
; (b) L. O. Alimi, D. P. van Heerden, P. Lama, V. J. Smith and L. J. Barbour, Chem. Commun., 2018, 54, 6208 RSC
-
(a) B. Sandhu, A. S. Sinha, J. Desper and C. B. Aakeröy, Chem. Commun., 2018, 54, 4657 RSC
- A. Forni, P. Metrangolo, T. Pilati and G. Resnati, Cryst. Growth Des., 2004, 4, 291 CrossRef
- K. M. Hutchins, K. A. Kummer, R. H. Groeneman, E. W. Reinheimer, M. A. Sinnwell, D. C. Swenson and L. R. MacGillivray, CrystEngComm, 2016, 18, 8354 RSC
- S. Bhattacharya and B. K. Saha, Cryst. Growth Des., 2013, 13, 3299 CrossRef
-
(a) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478 CrossRef PubMed
- C. M. Santana, E. W. Reinheimer, H. R. Krueger Jr., L. R. MacGillivray and R. H. Groeneman, Cryst. Growth Des., 2017, 17, 2054 CrossRef
- K. M. Hutchins, D. K. Unruh, F. A. Verdu and R. H. Groeneman, Cryst. Growth Des., 2018, 18, 566 CrossRef
-
(a) A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514 CrossRef PubMed
- M. J. Cliffe and A. L. Goodwin, J. Appl. Crystallogr., 2012, 45, 1321 CrossRef
- B. K. Saha, J. Indian Inst. Sci., 2017, 97, 177 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, single-crystal X-ray data, intermolecular interaction distances, and PASCal thermal expansion results. CCDC 1850139–1850153. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ce01090k |
| ‡ Due to issues with crystal decay during data collection at 290 K for (C6Br2F4)·(4,4′-BPA), we were not able to get a quality X-ray crystal structure. All TE parameters are based on 270 K as the highest temperature. |
| § (C6Br4H2)·(4,4′-BPA) was determined to be isostructural with the previously reported co-crystal (C6Br4H2)·(4,4′-BPE).7 |
| This journal is © The Royal Society of Chemistry 2018 |