
Time modulation of defects in UiO-66 and application in oxidative desulfurization†
Weiming
Xiao
 *,
Qiaoling
Dong
,
Yao
Wang
,
Yuan
Li
,
Shengjun
Deng
and
Ning
Zhang
*
*,
Qiaoling
Dong
,
Yao
Wang
,
Yuan
Li
,
Shengjun
Deng
and
Ning
Zhang
*
Institute of Applied Chemistry, College of Chemistry, Nanchang University, Nanchang, Jiangxi 330031, P. R. China. E-mail: xiaoweiming@ncu.edu.cn; nzhang.ncu@163.com
First published on 15th August 2018
Abstract
A defective porous metal–organic framework, UiO-66, prepared by simply tuning the synthesis time, shows excellent efficiency for oxidative desulfurization.
Metal–organic frameworks (MOFs) have been considered very promising heterogeneous catalysts owing to their large surface area and high density of active sites as well as having highly tunable properties.1 UiO-type materials are a class of MOFs based on Zr6O4(OH)4(CO2)12 secondary building units (Zr6 SBUs).2 The exceptional metal–ligand bond strength in Zr6 SBUs endows UiO-type MOFs with high mechanical, thermal and chemical stability,3 which has made such MOFs suitable for many applications including chemical sensing, sorption and catalysis.4 Among these UiO-type frameworks, UiO-66 using terephthalic acid (BDC) as a linker has received the most attention due to its relatively low cost, high stability and large surface area.5
In catalysis, UiO-66 is commonly used as a solid support, while few studies explore its intrinsic catalytic properties.6 This is because pristine UiO-66 lacks catalytic active sites. To enable the application of UiO-66, defect engineering has been reported to be a promising approach. The defects can liberate Lewis acidic sites (Zr4+ sites with unsaturated coordination) and lead to a more open framework in UiO-66, which provides novel opportunities for applications in catalysis.7 To date, numerous works have reported the tuning of the defects of UiO-66. In most cases, the procedures refer to replacing the BDC linkers with monocarboxylic acids, termed modulators and linker fragments.8 Only a few reports tuned the defects of UiO-66 by varying the molar ratio of linker to Zr-source or synthesis temperature.5c,9 These defect engineering methods always need long reaction times (>18 h).
The sulfur compounds in petroleum-based fuel cause catalyst poisoning and corrosion of equipment during crude oil processing. They also result in the emission of harmful SOx gases after combustion. Sulfur content limits are becoming increasingly stringent worldwide, especially for gasoline and diesel.10 To remove sulfur compounds from fuel, hydrodesulfurization (HDS) has been commonly used due to its efficiency in the elimination of mercaptans, sulfides and disulfides. However, it is less effective at the removal of refractory sulfur compounds such as dibenzothiophene (DBT) and its derivatives. In this context, cost-effective methods that are complementary to HDS for producing sulfur-free fuels have been developed.11 Oxidative desulfurization (ODS), as one of the most promising processes, has received considerable attention due to its efficiency and low cost. In ODS, refractory sulfur molecules are oxidized to their corresponding polar molecules, which can be removed from fuel by simple solvent extraction.12
By considering these points, we wish to report a facile method to create defects in UiO-66. By simply tuning the synthesis time, a series of defective UiO-66 materials were obtained and their catalytic activities in ODS were also examined. The synthesis of MOFs generally involves the formation and dissolution of nuclei, where the synthesis time plays a significant role. Therefore, controlling the synthesis time will tune the formation of the MOFs. In the current work, UiO-66 with defects was prepared by stirring a mixture of ZrCl4, terephthalic acid, aqueous NH3and DMF at 110 °C for different reaction times. Then, the products were separated by centrifugation and washed with DMF and methanol and denoted UiO-66-t (t stands for synthesis time and the time unit is hours). Details of the synthesis are presented in the ESI.† The XRD patterns suggest a highly crystalline nature and are in good agreement with the simulated pattern reported in the literature,5a revealing the successful synthesis of UiO-66 (Fig. 1a). The full width at half maximum (FWHM) values of the samples were calculated. Almost all the FWHM values of the selected diffraction peaks decrease with increasing synthesis time (Fig. S1†), which indicates that the crystallite size of the as-synthesized UiO-66 increases with synthesis time according to the Scherrer equation. The crystallite size is generally considered inversely proportional to the defect concentration in crystalline solids.13 Thus, a fast synthesis of UiO-66 may result in more defects in the framework. Moreover, the growth of the UiO-66 is also observed in the SEM images, where the average particle size increases with synthesis time from ca. 15 nm to ca. 210 nm (Fig. S2 and Table S1†), which further confirms the XRD results.
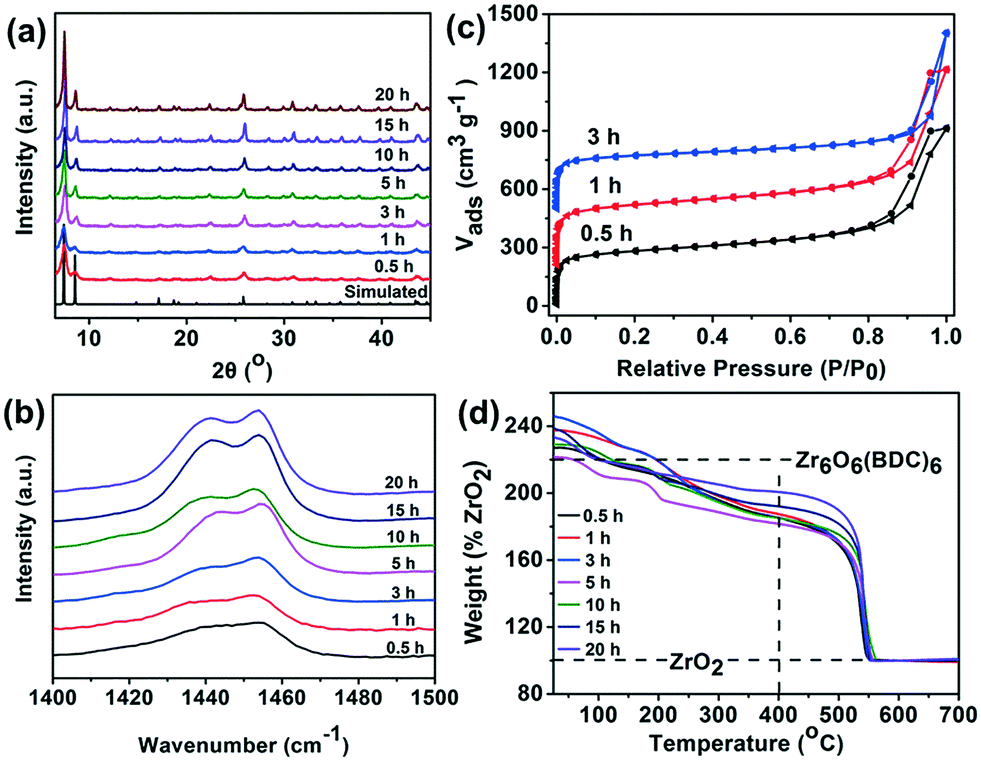 | ||
| Fig. 1 Characterization of UiO-66 with different synthesis times. (a) XRD patterns, (b) Raman spectra and (c) N2 adsorption–desorption isotherms at 77 K. For clarity, the isotherms of UiO-66-1 h and UiO-66-3 h are offset along the y axis by 200 and 500 cm3 g−1, respectively. (d) TGA profiles. | ||
Raman spectroscopy was also employed to confirm the defects in the UiO-66-t samples. The bands located at 1474–1420 cm−1 are associated with carboxylate-related stretches, which provide information about the linkers missing from UiO-66.2,5c For ideal UiO-66, two distinct bands can be observed in this range. In Fig. 1b, the band around 1450 cm−1 is gradually separated into two bands with increasing synthesis time, indicating the reduction of linker defects within the frameworks of UiO-66 samples with long synthesis time.
The porous structures of the UiO-66-t samples were evaluated using the N2 sorption isotherms at 77 K (Fig. 1c and S3†). The UiO-66-t samples show large specific surface areas, all exceeding 1000 m2 g−1. In contrast to pristine UiO-66, which features a microporous structure, the UiO-66-t, especially the quickly synthesized samples (UiO-66-0.5 h, UiO-66-1 h, UiO-66-3 h) exhibit type-IV isotherms with an H3-type hysteresis loop between relative pressure P/P0 = ∼0.7–1.0, indicating the presence of mesopores in the quickly synthesized samples. The pore size distributions obtained from a non-local density functional theory model also suggest the broad mesopore distributions in the UiO-66-t samples (Fig. S4†). The formation of mesopores in microporous MOFs via tuning the synthesis time has also been observed by S. Dai's group.14 Table S2† summarizes the pore parameters for the UiO-66-t samples. With long synthesis times, the mesopores decrease while the micropores greatly increase. Generally, the large pores usually contain abundant defects.7d,15 This indicates that the quickly synthesized UiO-66 samples probably possess a high concentration of defects, which is in accordance with the XRD and Raman results.
TGA is one of the most accessible methods that can provide information about missing linker defects in MOFs,16 although it has come under criticism because the assessment only considers missing linkers but not missing inorganic bricks in the framework.15,17 For perfect UiO-66, each Zr6O4(OH)4 core connects to 12 linkers, while one linker bridges two metal cores, thus the structural unit is Zr6O4(OH)4(BDC)6. Upon heat treatment, the removal of solvents and dehydroxylation of the Zr6 cluster are completed at ca. 400 °C, giving an inner cluster of Zr6O6(BDC)6.2,5b,15 The decomposition of the BDC linker starts at ca. 450 °C, finally forming ZrO2 in air. If the end weight is normalized as 100%, the weight around 400 °C for the Zr6O6(BDC)6 should ideally reach 220%. The TGA curves of the UiO-66-t samples are presented in Fig. 1d. The normalized weight around 400 °C for all the UiO-66-t samples is clearly below the expected value (220%). The apparent concentrations of the missing linker defects in UiO-66-0.5 h, UiO-66-1 h, UiO-66-3 h, UiO-66-5 h, UiO-66-10 h, UiO-66-15 h and UiO-66-20 h are estimated to be 32%, 33%, 35%, 38%, 35%, 28% and 19%, respectively. These values are probably inaccurately estimated due to the fact that missing inorganic brick defects likely coexist in the synthesized UiO-66.15,17 However, it still indicates that the quickly synthesized UiO-66 samples possess abundant missing linker defects.
More missing linker defects can create more acid sites in UiO-66. NH3-TPD is an effective method for identifying the acid content in solid materials. Thus, NH3-TPD was carried out in this work. As shown in Fig. S5,† the NH3 desorption of UiO-66-0.5 h is higher than that of UiO-66-15 h, revealing the higher acid concentration of UiO-66-0.5 h. This further proves that rapid synthesis results in more defects in UiO-66.
An amorphous phase may be formed in the UiO-66 samples with a high concentration of defects.18 However, only traces of amorphous phases are observed in the as-synthesized UiO-66 samples, considering the intensive peaks of the XRD patterns and Raman spectra (Fig. 1a and S6†). Thus, these findings can conclude that the defective UiO-66 samples were successfully prepared via tuning the synthesis time and that rapid synthesis offers abundant defects in UiO-66.
In ODS, H2O2 has been widely used as the oxidant owing to its environmental friendliness and low cost.19 The defects liberate unsaturated Zr4+ sites in UiO-66, which can interact with H2O2 to form Zr-peroxo complexes.20 The Zr-peroxo groups are very active in the oxidation of sulfur substrates to their corresponding oxide products2,21 (Scheme 1). In this work, the oxidation of DBT using UiO-66-t was studied in acetonitrile. As shown in Fig. 2a, all the UiO-66-t samples exhibit catalytic activity and their activities decrease expectedly with increasing synthesis time. The UiO-66-0.5 h and UiO-66-1 h display the best activity, with nearly complete oxidation of DBT after only 20 min of reaction time. Moreover, the current work also investigated the catalytic performance of UiO-66 prepared using a solvothermal method (named UiO-66-S). UiO-66-S has a similar particle size but fewer defects as compared with the UiO-66-5 h (Fig. S2 and S7†), while its activity is significantly lower than that of UiO-66-5 h (Fig. S8†). This indicates that the defects in UiO-66 play a more important role in the oxidation of DBT. Thus, the high activities of UiO-66-0.5 h and UiO-66-1 h can be ascribed to their highly defective structures.
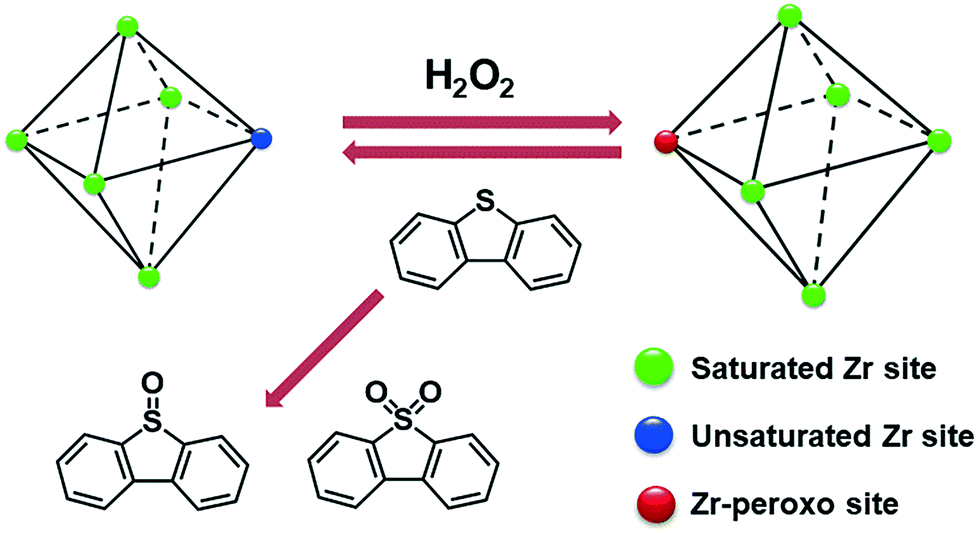 | ||
| Scheme 1 The reaction of DBT oxidation using defective UiO-66 as a catalyst and H2O2 as an oxidant. | ||
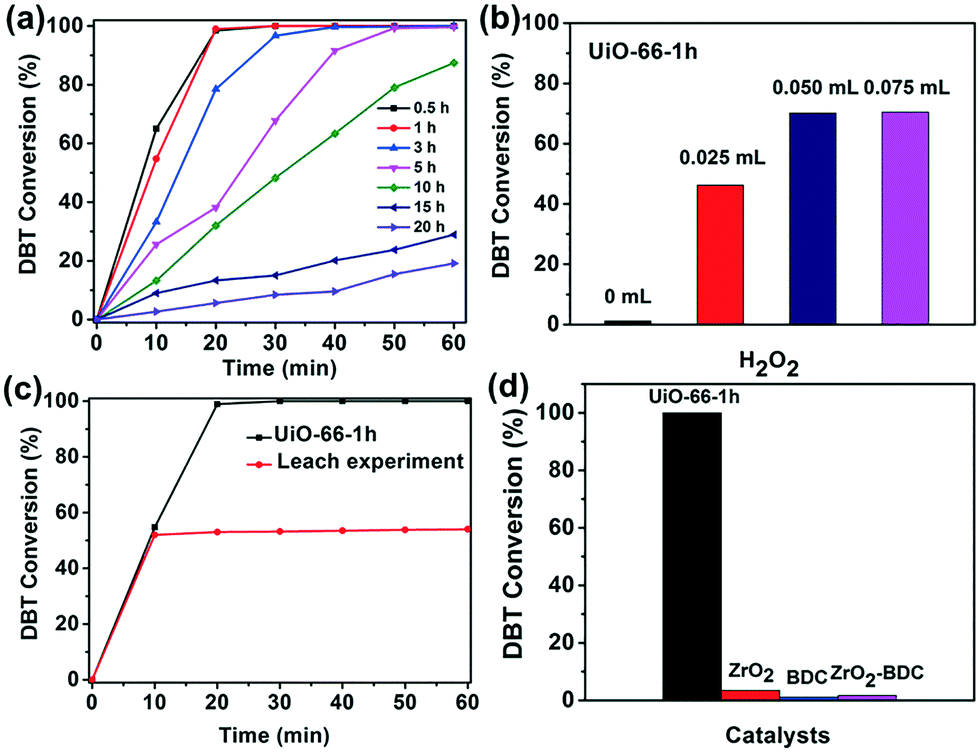 | ||
| Fig. 2 (a) Catalytic activities of UiO-66 with different synthesis times (H2O2: 0.05 mL). (b) Effect of H2O2 amount (time: 15 min). (c) Leaching test (H2O2: 0.05 mL). (d) Control experiments (H2O2: 0.05 mL, time: 60 min). Catalyst: 5 mg; temperature: 60 °C; DBT in acetonitrile: 2 mL (1000 ppm of sulfur). | ||
Optimization of the usage of H2O2 was carried out with UiO-66-1 h (Fig. 2b). After a 15 min reaction, the oxidation of DBT hardly happens without H2O2. The oxidation process using 0.05 ml of aqueous H2O2 shows a notably higher oxidation efficiency (70%) than that with 0.025 ml (46%). Upon further increasing the amount of H2O2 to 0.075 ml, no significant improvement in oxidation efficiency is observed. For this, 0.05 ml of aqueous H2O2 was selected for this oxidation reaction. The heterogeneity of UiO-66-t was also investigated. The leaching test was performed on the UiO-66-1 h. The catalyst was separated from the mixture after a 10 min reaction and the reaction continued with the remaining filtrate. As shown in Fig. 2c, the oxidation of DBT almost immediately stops after the catalyst is removed, confirming the heterogeneity. For comparison, control experiments were carried out using ZrO2, BDC and a ZrO2–BDC mixture as the catalysts (Fig. 2d). The results show that all of these control catalysts exhibit nearly no activity for the oxidation reaction, which indicates that the activities of the UiO-66-t samples do not originate from the possible presence of ZrO2 and uncoordinated BDC.
Since UiO-66-1 h shows excellent catalytic activity for the oxidation of DBT, its activity for the desulfurization of a model oil containing DBT was evaluated. Fig. 3a suggests that UiO-66-1 h exhibits a high efficiency for the removal of DBT in n-octane, with 97% DBT removal after a 1 h reaction. The reusability of UiO-66-1 h for the ODS reaction was also investigated. At the end of the reaction, the catalyst was separated by centrifugation, washed with methanol and dried at 80 °C, then used again. As shown in Fig. 3b, UiO-66-1 h shows poor recyclability. The sulfur removal shows significant decrease from 97% to 68% after only one run. XRD and N2 sorption were used to examine the stability of the UiO-66-1 h. The results demonstrate that the reused UiO-66-1 h has a similar structure to the fresh one (Fig. S9†), indicating the stability of UiO-66-1 h. Therefore, the deactivation of UiO-66-1 h is not caused by the collapse of UiO-66-1 h. The reason for the deactivation is probably due to the strong adsorption of sulfone (oxidized product of DBT) on the active sites of UiO-66-1 h. This phenomenon has also been observed by M. Zhu's group.22
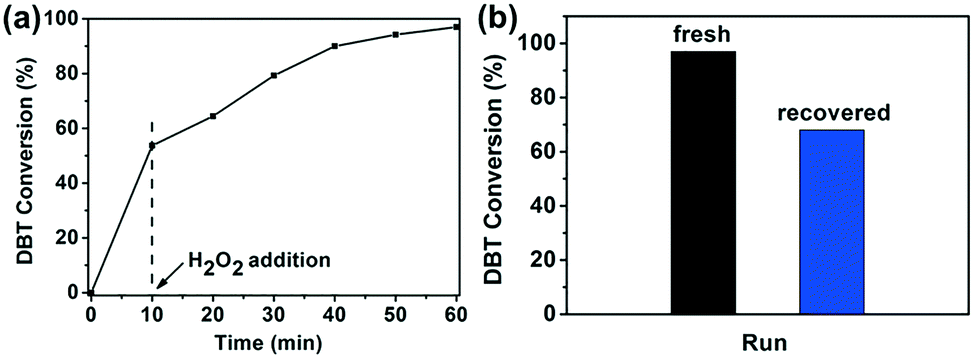 | ||
| Fig. 3 (a) Catalytic profile of UiO-66-1 h for ODS. (b) Performance of the recovered UiO-66-1 h (time: 60 min). Catalyst: 50 mg; H2O2: 0.5 mL; DBT in n-octane (1000 ppm sulfur): 20 mL; acetonitrile: 20 mL; temperature: 60 °C. | ||
Conclusions
In summary, this work presents a facile method for creating defects in UiO-66 via tuning the synthesis time. The structural analysis reveals that fast synthesis facilitates the formation of defects in UiO-66, and the defects are beneficial for the catalysis. The quickly synthesized samples UiO-66-0.5 h and UiO-66-1 h possess a high concentration of defects and show excellent performance in the oxidation of DBT. We believe that this time modulation method can inspire more work on the design of advanced defective MOFs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financed by the National Natural Science Foundation of China (No. 21263014 and 21663016), and the Natural Science Foundation of Educational Department of Jiangxi Province (No. GJJ150083).Notes and references
-
(a) H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef
; (b) L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC
- C. M. Granadeiro, S. O. Ribeiro, M. Karmaoui, R. Valenca, J. C. Ribeiro, B. de Castro, L. Cunha-Silva and S. S. Balula, Chem. Commun., 2015, 51, 13818–13821 RSC
-
(a) V. Guillerm, F. Ragon, M. Dan-Hardi, T. Devic, M. Vishnuvarthan, B. Campo, A. Vimont, G. Clet, Q. Yang, G. Maurin, G. Ferey, A. Vittadini, S. Gross and C. Serre, Angew. Chem., Int. Ed., 2012, 51, 9267–9271 CrossRef PubMed
-
(a) C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef PubMed
-
(a) J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed
-
(a) R. Wu, X. Qian, K. Zhou, H. Liu, B. Yadian, J. Wei, H. Zhu and Y. Huang, J. Mater. Chem. A, 2013, 1, 14294–14299 RSC
-
(a) F. Vermoortele, B. Bueken, G. Le Bars, B. Van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. Van Speybroeck, C. Kirschhock and D. E. De Vos, J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef PubMed
-
(a) Y. Liu, R. C. Klet, J. T. Hupp and O. Farha, Chem. Commun., 2016, 52, 7806–7809 RSC
- M. R. DeStefano, T. Islamoglu, S. J. Garibay, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 1357–1361 CrossRef
-
(a) A. Bösmann, L. Datsevich, A. Jess, A. Lauter, C. Schmitz and P. Wasserscheid, Chem. Commun., 2001, 2494–2495 RSC
-
(a) C. Song, Catal. Today, 2003, 86, 211–263 CrossRef
-
(a) F. Li, R. Liu, J. Wen, D. Zhao, Z. Sun and Y. Liu, Green Chem., 2009, 11, 883–888 RSC
-
(a) A. A. Baig, J. L. Fox, R. A. Young, Z. Wang, J. Hsu, W. I. Higuchi, A. Chhettry, H. Zhuang and M. Otsuka, Calcif. Tissue Int., 1999, 64, 437–449 CrossRef PubMed
- Y. Yue, Z. A. Qiao, P. F. Fulvio, A. J. Binder, C. Tian, J. Chen, K. M. Nelson, X. Zhu and S. Dai, J. Am. Chem. Soc., 2013, 135, 9572–9575 CrossRef PubMed
- L. Yuan, M. Tian, J. Lan, X. Cao, X. Wang, Z. Chai, J. K. Gibson and W. Shi, Chem. Commun., 2018, 54, 370–373 RSC
- Z. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54, 7234–7254 CrossRef PubMed
- M. J. Cliffe, W. Wan, X. Zou, P. A. Chater, A. K. Kleppe, M. G. Tucker, H. Wilhelm, N. P. Funnell, F. X. Coudert and A. L. Goodwin, Nat. Commun., 2014, 5, 4176 CrossRef PubMed
-
(a) T. D. Bennett, A. K. Cheetham, A. H. Fuchs and F. X. Coudert, Nat. Chem., 2017, 9, 11–16 CrossRef PubMed
-
(a) J. L. García-Gutiérrez, G. A. Fuentes, M. E. Hernández-Terán, F. Murrieta, J. Navarrete and F. Jiménez-Cruz, Appl. Catal., A, 2006, 305, 15–20 CrossRef
-
(a) M. T. H. Tarafder and M. A. L. Miah, Inorg. Chem., 1986, 25, 2265–2268 CrossRef
-
(a) E. Torres-García, A. Galano and G. Rodriguez-Gattorno, J. Catal., 2011, 282, 201–208 CrossRef
- X. Zhang, P. Huang, A. Liu and M. Zhu, Fuel, 2017, 209, 417–423 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supporting figures and tables. See DOI: 10.1039/c8ce00795k |
| This journal is © The Royal Society of Chemistry 2018 |