
Controlled synthesis of t-Se nanomaterials with various morphologies via a precursor conversion method
Jianhua
Zhang
 ,
Qingshan
Fu
,
Yongqiang
Xue
* and
Zixiang
Cui
,
Qingshan
Fu
,
Yongqiang
Xue
* and
Zixiang
Cui
Department of Applied Chemistry, Taiyuan University of Technology, Taiyuan 030024, P R China. E-mail: xyqlw@126.com; Tel: +86 351 6014476
First published on 17th January 2018
Abstract
Trigonal selenium (t-Se) nanomaterials with different morphologies present distinct properties and great potential applications in electric devices. However, controlled synthesis of t-Se nanomaterials with various morphologies is difficult in a typical preparation process. Therefore, it is imperative to develop an easily controlled and high-efficiency method to prepare t-Se with various morphologies. Herein, a precursor conversion method was proposed to prepare t-Se nanomaterials with different morphologies. That is, uniform amorphous selenium (a-Se) nanospheres were prepared by reducing sodium selenite with glucose, and then t-Se nanomaterials with morphologies of spheres, tubes, rods, belts and wires were obtained by different subsequent treatments for the conversion of a-Se into t-Se. The results demonstrate that the t-Se nanospheres were obtained by hydrothermal treatment at 150 °C, t-Se nanorods and nanotubes by ultrasonication of a-Se in water and with the addition of PVP K30 for nanotubes, t-Se nanowires by the aging of a-Se in ethanol and in a dark environment, and t-Se nanobelts by increasing the concentration of a-Se in ethanol. The conversion processes from a-Se nanospheres into t-Se 1D nanostructures comply with a “solid–solution–solid” formation mechanism, while the conversion from a-Se nanospheres into t-Se nanospheres complies with the mechanism of crystalline phase transformation. The method provides us a mild and easily controlled route for the preparation of t-Se nanomaterials with desired morphologies.
1. Introduction
Recently, t-Se nanomaterials have attracted considerable attention due to their unique optical, electrical and magnetic properties, which can be used as basic components in the fabrication of nano-sized electric devices, such as xerography, electrical rectifier meters, mechanical sensors, photocells,1–10etc. In addition, due to its high reactivity towards a number of chemicals, selenium with special morphologies can serve as a template to be transformed into many other functional materials, such as RuSe2,11 AgSe,12 ZnSe,13 CdSe,14 PbSe,15etc. These excellent properties and special applications of t-Se nanomaterials are closely related to their morphology, and hence, searching for a mild and easily controlled method to synthesize t-Se nanostructures with different morphologies has important scientific significance for their applications.Several methods on the preparation of t-Se with different morphologies have been reported. Xie et al.16 synthesized selenium microtubes, nanorods, shuttle-like needles, and urchin-like assemblies of selenium nanorods based on irradiation with visible light from a commercial lawn lamp. Goia et al.17 prepared t-Se spherical particles, wires and rods by reducing selenous acid with hydroquinone in the presence of Daxad 11G. Quan et al.18 obtained selenium nanowires, nanorods, nanotubes and nanobelts based on a solution-mediated heat treatment with commercially available Se powders. Mondal and Srivastava19 produced single crystalline t-Se with several morphologies (wires, rods, flowerlike and hollow spheres) by hydrothermal reaction. Although the above methods can successfully synthesize t-Se with different morphologies, the preparation method of t-Se with various morphologies by a precursor conversion method for converting a-Se into t-Se has not been reported.
Selenium is known to exist mainly in four allotropic forms including amorphous, trigonal, α-and β-monoclinic allotropes, of which the trigonal phase is the most stable crystal form and amorphous selenium can spontaneously transform into the trigonal phase under appropriate conditions. Based on this, a precursor conversion method was proposed to prepare trigonal selenium with various morphologies. In our work, a-Se was firstly prepared by reducing sodium selenite with glucose, and then t-Se nanomaterials with various morphologies (selenium nanospheres, nanotubes, nanorods, nanoribbons and nanowires) were synthesized through the conversion process of a-Se into the trigonal phase (t-Se) and by altering the concentration of a-Se in solution, the kinds of solvents, the sonication and the surfactant. Finally, the formation mechanisms of t-Se nanomaterials with various morphologies were discussed.
2. Experimental section
2.1. Materials
Glucose, sodium selenite and anhydrous ethanol were purchased from Tianjin Guangfu Fine Chemical Research Institute, PVP K30 was purchased from Aladdin, all of the chemical reagents in our experiments were of analytical grade and used without further purification, and the water used throughout the experiment was purified using a Milli-Q water purification system.2.2. Preparation of a-Se nanospheres and t-Se nanospheres
0.25 g sodium selenite and 1.5 g glucose were dissolved in 100 mL distilled water, and then the solution was heated in a water bath for 20 min without stirring at 90 °C, which was accompanied by a gradual color change from colorless to brick red. After the hot solution was cooled down to quench the reaction quickly, the a-Se nanospheres were separated from the solution by centrifugation, washed with deionized water and alcohol, and dried in a vacuum oven for the following characterization and determination. As for the t-Se nanosphere synthesis, the same procedure was used for preparing the solution, and then the solution was transferred into a Teflon-lined stainless steel autoclave with a capacity of 150 mL and kept in an oven at 150 °C for 5 h without stirring.2.3. Preparation of t-Se nanowires and nanobelts
The as-prepared a-Se (0.04 g) was dispersed (sonication for 15 to 25 s (ref. 20)) in 100 mL absolute ethanol, and then the dispersion was aged for one week in a dark environment at room temperature. Finally, black-gray t-Se nanowires were precipitated at the bottom of the beaker. The t-Se nanobelts were obtained by increasing the concentration of a-Se in ethanol.2.4. Preparation of t-Se nanorods and t-Se nanotubes
The as-prepared 5 mg a-Se was ultrasonically dispersed in 20 mL distilled water under sonication for several hours; the selenium nanorods were obtained after the brick red amorphous selenium became black-gray. As for the synthesis of nanotubes, the same procedure was used except for the addition of 0.5 g of PVP K30 into the initial reaction mixture at room temperature. The frequency, the power, and the instrument model adopted in this sonochemical synthesis is 59 kHz, 250 W cm−2 and Kudos (SK250LH), respectively.2.5. Characterization
Powder X-ray diffraction (PXRD) measurements were performed on a Bruker D8 Advance X-ray diffractometer (XRD) with Cu KR radiation in the 2θ range from 20 to 80°. SEM images were obtained using a field emission scanning electron microscope (JSM-7001F). TEM measurements were performed on a Philips CM 20 FEG 200 keV transmission electron microscope. Raman spectra were recorded on a Spex 1403 Raman spectrometer using a 514 nm laser beam. Thermal analysis was performed on a differential scanning calorimeter (STA 449F3).3. Results and discussion
3.1. Nanospheres of a-Se and t-Se
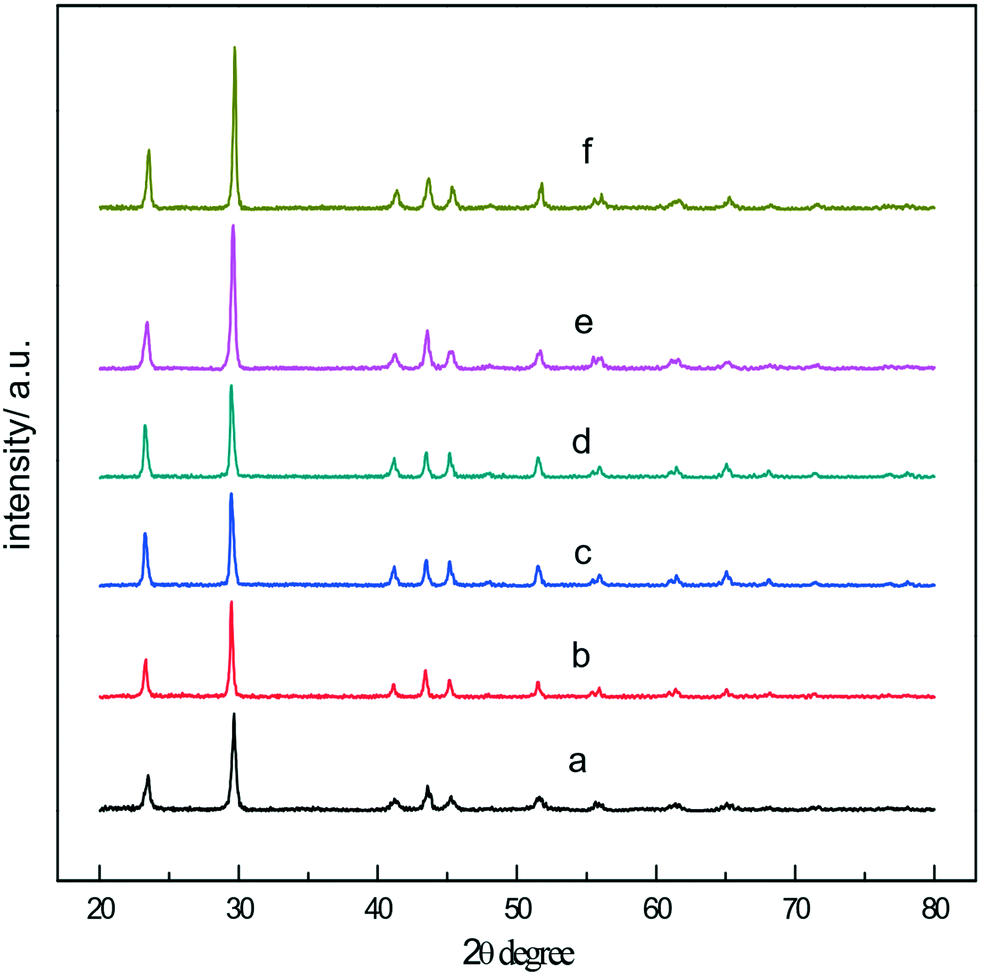
Fig. 1a and b are the SEM images of the a-Se products at different magnifications, and it can be seen that the product is composed of uniform spherical nanoparticles with a diameter of about 300 nm. Fig. 1c shows the energy dispersive spectra (EDS) for the a-Se products. In the EDS, selenium is the only element detected, showing that the sample is highly pure. A very broad feature in the XRD pattern (Fig. 1d) indicates that the product is amorphous selenium (a-Se) in nature.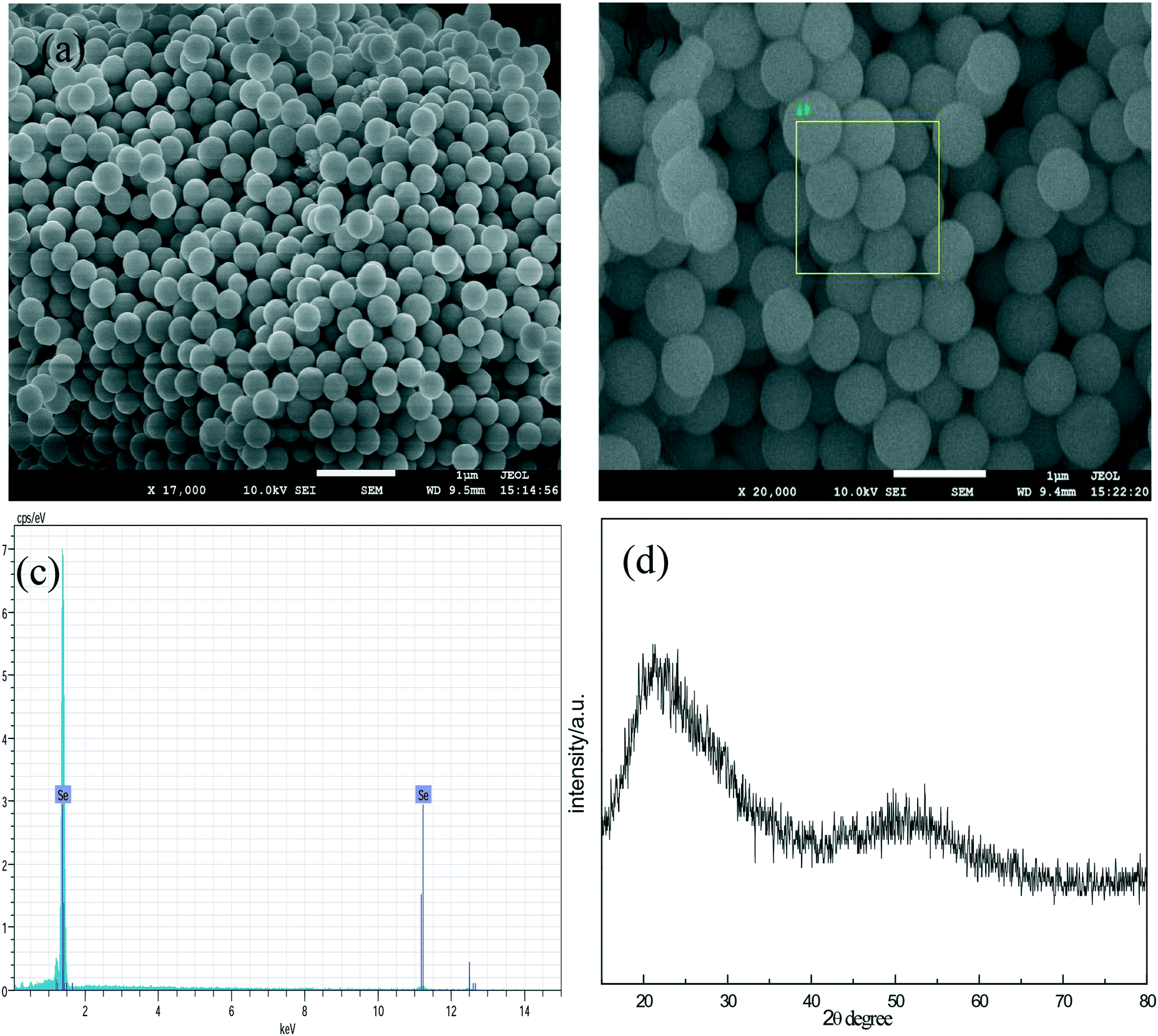 | ||
| Fig. 1 (a) and (b) SEM images of a-Se; (c) EDS of a-Se; (d) XRD of a-Se. | ||
In addition, we also found that the reactant concentrations have a significant effect on the diameter of amorphous selenium; the diameter of a-Se increases with the increase in the dosage of glucose and sodium selenite. This may be because the higher the amount of reactant dosage, the more the a-Se nuclei were reduced in the solution; the particle growth is overwhelmingly faster than the nucleation process at higher concentrations, which results in larger particle size. It is worth noting that the color of solution can change into dark red when the reaction time was prolonged to 2 hours; the presence of a dark red color indicates that some a-Se has been converted into t-Se, and hence, the reaction time is a key factor for the preparation of a-Se.
Fig. 2 shows the DSC curve of the amorphous selenium with a diameter of 300 nm heated at 10 °C min−1. An exothermic peak followed by an endothermic one is shown in the DSC curve. The exothermic peak is derived from a phase transformation from the amorphous selenium to the trigonal phase, and the endothermic peak corresponds to a melting process of the t-Se. The phase transition temperature of a-Se to t-Se is 123 °C. Namely, the a-Se can convert into t-Se when the temperature is above the phase transformation temperature; if the phase change can be completed quickly, the t-Se will maintain the original morphology of the a-Se.
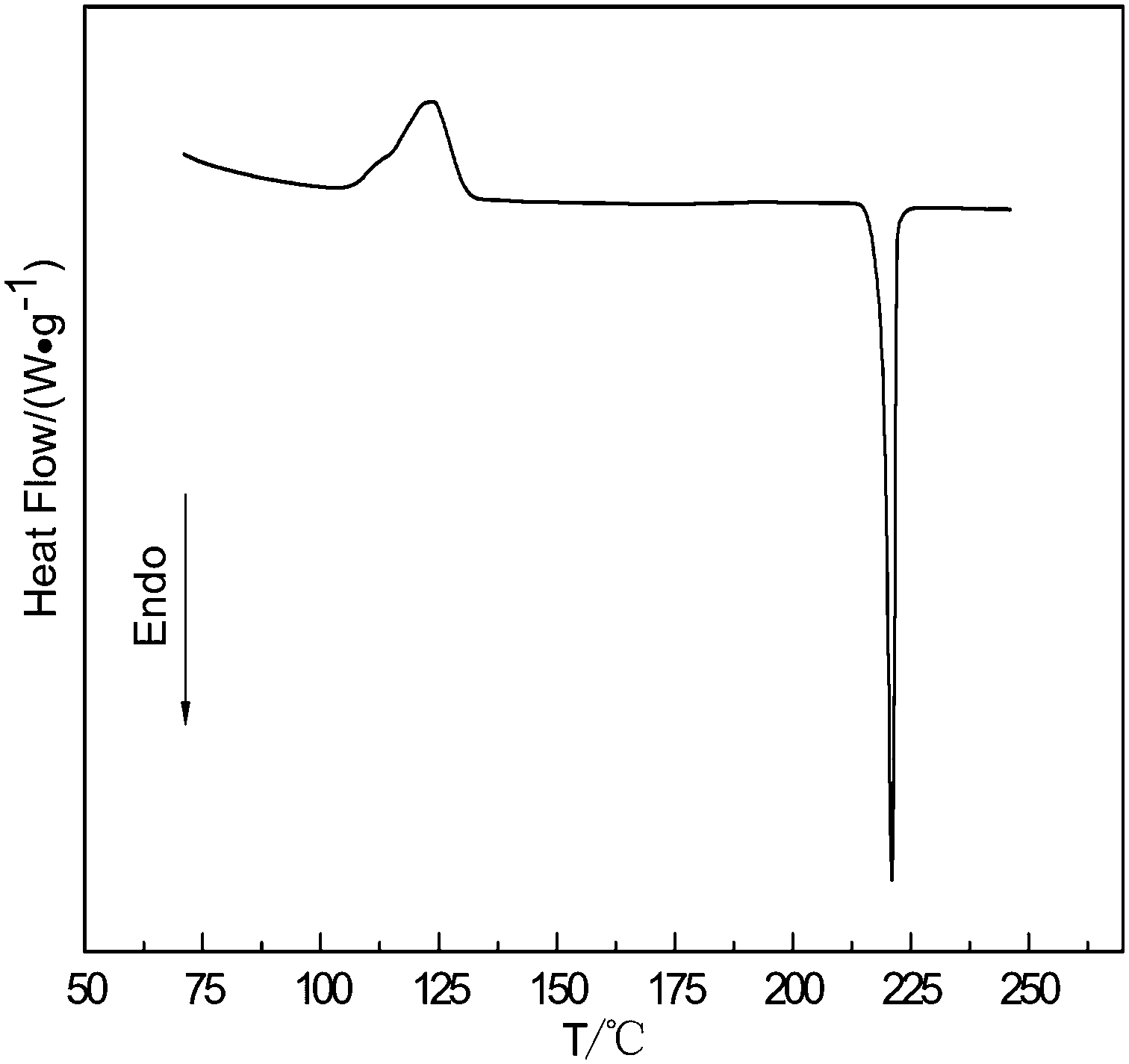 | ||
| Fig. 2 DSC curve of the amorphous selenium heated at a rate of 10 °C min−1. | ||
According to the principle of fast phase transition of a-Se at high temperature, t-Se nanospheres were obtained by hydrothermal reaction at 150 °C. Fig. 3a shows the SEM image of the t-Se product which is composed of monodisperse nanocrystals with a diameter of about 300 nm. As expected, the a-Se nanospheres and t-Se nanospheres almost have identical particle size, which confirms that the t-Se nanospheres were formed directly from a-Se nanospheres. The experimental results show that uniform t-Se nanospheres can be obtained when the hydrothermal temperature is between 140 and 180 °C. The hydrothermal reaction time was found to be 4–7 hours. Trigonal selenium was not generated when the hydrothermal reaction time was less than 2 hours, and the morphology of t-Se was non-uniform when the time was prolonged to 10 hours, which may be due to the presence of Ostwald ripening in the system. Moreover, the diameter of nanospheres increases with the increase in concentrations of sodium selenite and glucose. Fig. 3b is the SEM image of the t-Se after increasing the concentration of the reactants 2-fold, and it can be seen that the particle diameter of t-Se is about 550–600 nm, which agrees with the particle size in the TEM image (Fig. 3c). The XRD patterns of the two samples are presented in Fig. 3d, and all the diffraction peaks could be indexed to the t-Se (JCPDS 06-0362).
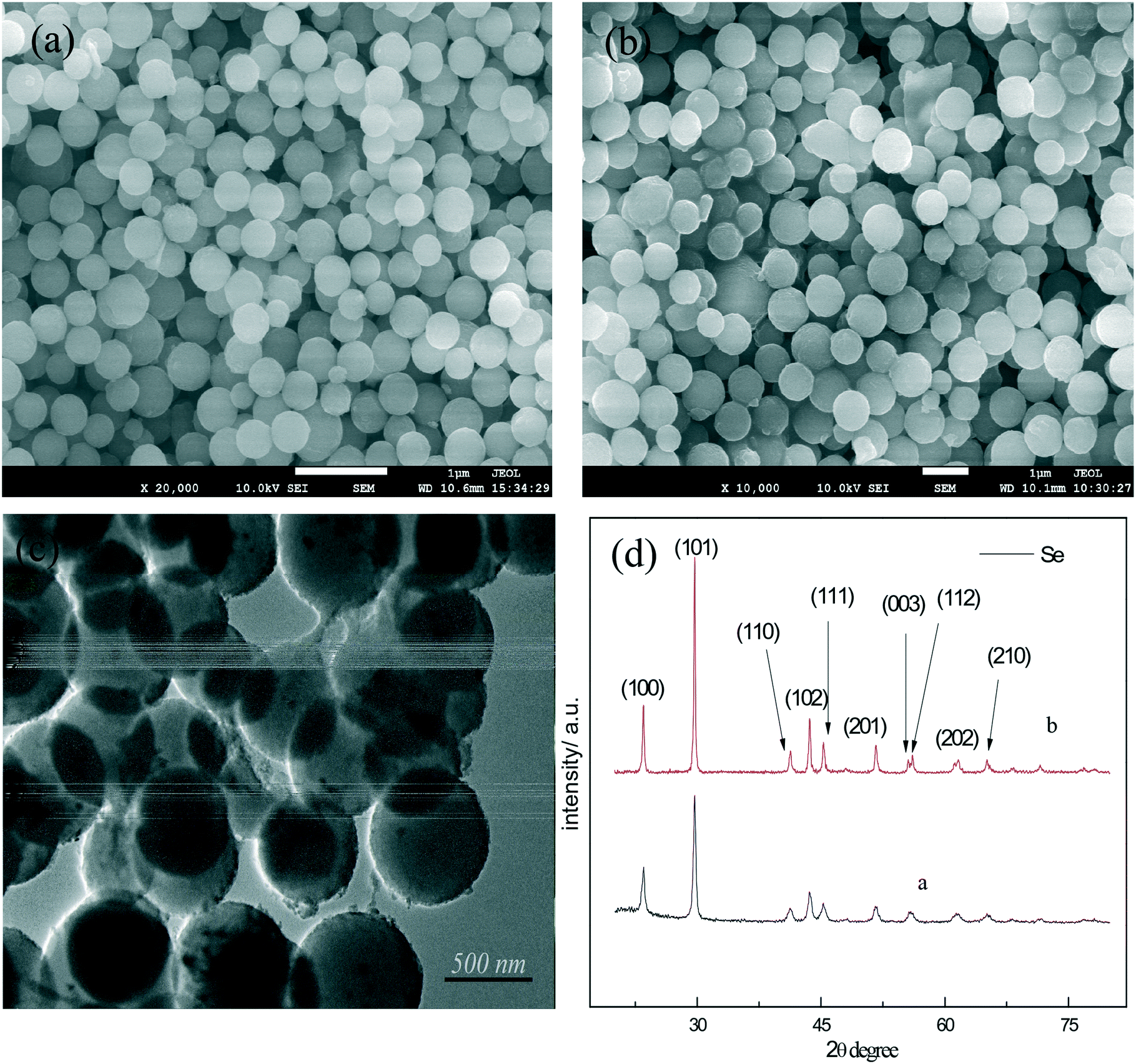 | ||
| Fig. 3 (a) SEM of t-Se; (b) SEM of t-Se obtained by increasing the concentration of the reactants 2-fold; (c) TEM of t-Se; (d) XRD patterns of the t-Se samples. Note: the letters in the XRD patterns correspond to the letters in the SEM images in this paper. | ||
3.2. Nanowires and nanobelts of t-Se
In this work, the t-Se nanowires were obtained after the a-Se aging in ethanol and in the dark environment for one week at room temperature. Fig. 4a shows the Raman spectrum of the as-obtained trigonal selenium nanowires. Only one resonance peak at 234 cm−1 is observed, without the appearance of the peaks of a-Se at 256 cm−1 and 264 cm−1 in the Raman spectrum, which indicates that the a-Se transformed completely into the t-Se. All the diffraction peaks in the XRD pattern in Fig. 4b match well with the trigonal phase of Se (JCPDS 06-0362). Fig. 4c shows the SEM image of t-Se nanowires at a low magnification in the typical synthesis, which displays a huge amount of t-Se nanowires with lengths up to several micrometers; the magnified SEM image in Fig. 4d shows that the diameter of these t-Se nanowires is about 40 nm, and each nanowire has a uniform diameter over the entire length. Fig. 4e shows a typical TEM image, from which we can find that the diameter of t-Se nanowires was almost consistent with the SEM result. The SAED pattern of t-Se nanowires was obtained by focusing the e-beam on an individual nanowire, and this pattern indicates that these t-Se nanowires have predominantly grown along the [001] direction.21,22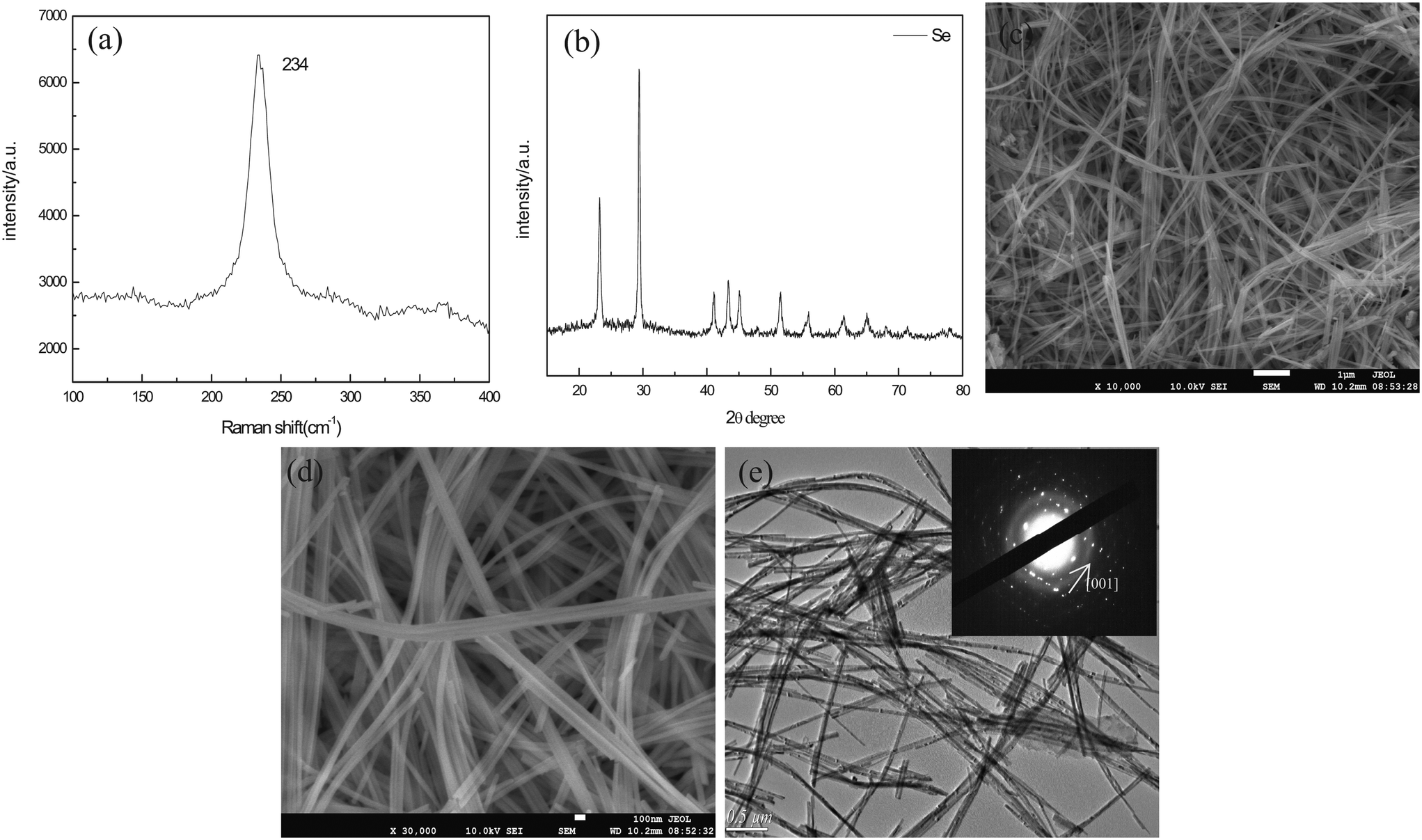 | ||
| Fig. 4 (a) Raman spectrum of t-Se; (b) XRD pattern of t-Se nanowires; (c) SEM image of t-Se nanowires at a low magnification; (d) SEM image of t-Se nanowires at a high magnification; (e) TEM image of t-Se nanowires, the insert shows the corresponding SAED pattern. | ||
The effects of aging time and concentration of a-Se on the growth of t-Se nanowires were studied. The morphology evolution of t-Se as a function of aging time is shown in Fig. 5. The morphology of the precursor (a-Se) was spherical originally (Fig. 5a), some short nanorods (Fig. 5b) were formed after the precursor was aged in absolute ethanol for 1 day and gradually grew into longer nanorods (Fig. 5c and d) after 3 and 5 days. The nanorods continue to grow and eventually turn into nanowires (Fig. 5e) after 7 days. The diameter of the initial a-Se nanospheres differs from that of the resulting t-Se nanowires, which indicates that the nanowires comply with a “solid–solution–solid” formation mechanism.23Fig. 6 illustrates a formation mechanism for the growth of nanowires. To begin with, the morphology of a-Se nanoparticles was spherical (Fig. 6a) and a small amount of a-Se nanoparticles converted spontaneously into t-Se seeds, due to the higher free energy of a-Se compared to that of t-Se (Fig. 6b); these t-Se seeds on the surface of a-Se nanoparticles grew along the c-axis ([001] direction) at the expense of a-Se nanoparticles owing to the intrinsically anisotropic structure of t-Se (Fig. 6c). The spontaneous growth process continued until all a-Se nanoparticles had been used up in the solution, eventually resulting in the formation t-Se nanowires (Fig. 6d). When the transformation of a-Se to t-Se was completed, further extension of the aging time could not obviously increase the length of the nanowires, which is consistent with the report of Chen et al.24
 | ||
| Fig. 5 SEM images of the a-Se aged in absolute ethanol for different numbers of days at room temperature: (a) precursor (a-Se); (b) 1 day; (c) 3 days; (d) 5 days; (e) 7 days. | ||
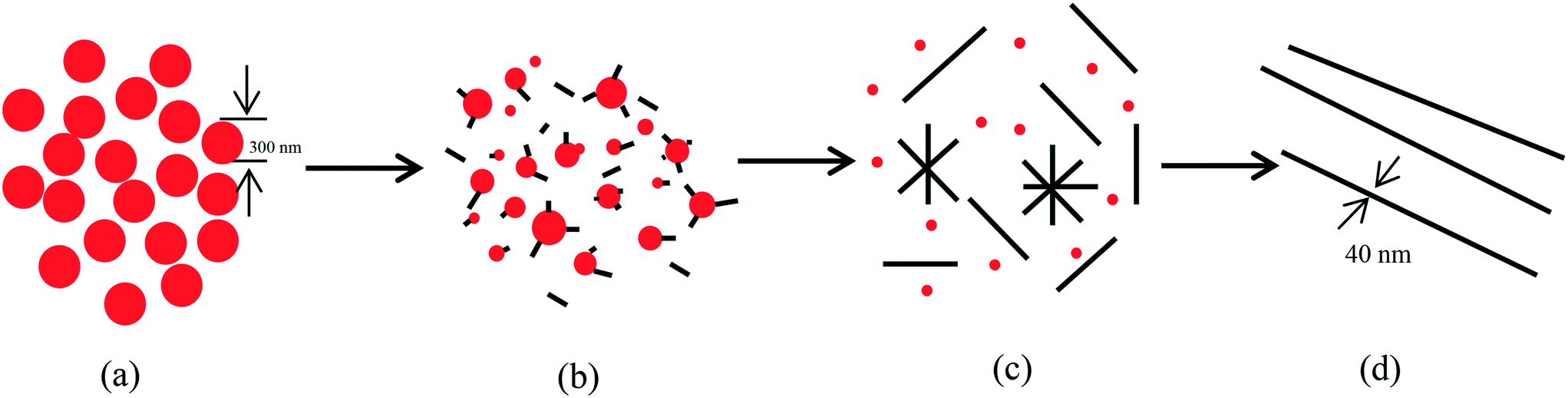 | ||
| Fig. 6 Schematic illustration of the formation of t-Se nanowires: (a) a-Se nanospheres; (b) formation of t-Se seeds on the surfaces of a-Se nanoparticles; (c) t-Se nanowires grown continually along the c-axis at the expense of the gradually dissolved a-Se nanoparticles; (d) t-Se nanowires. Note: the red color and gray color represent a-Se and t-Se, respectively. | ||
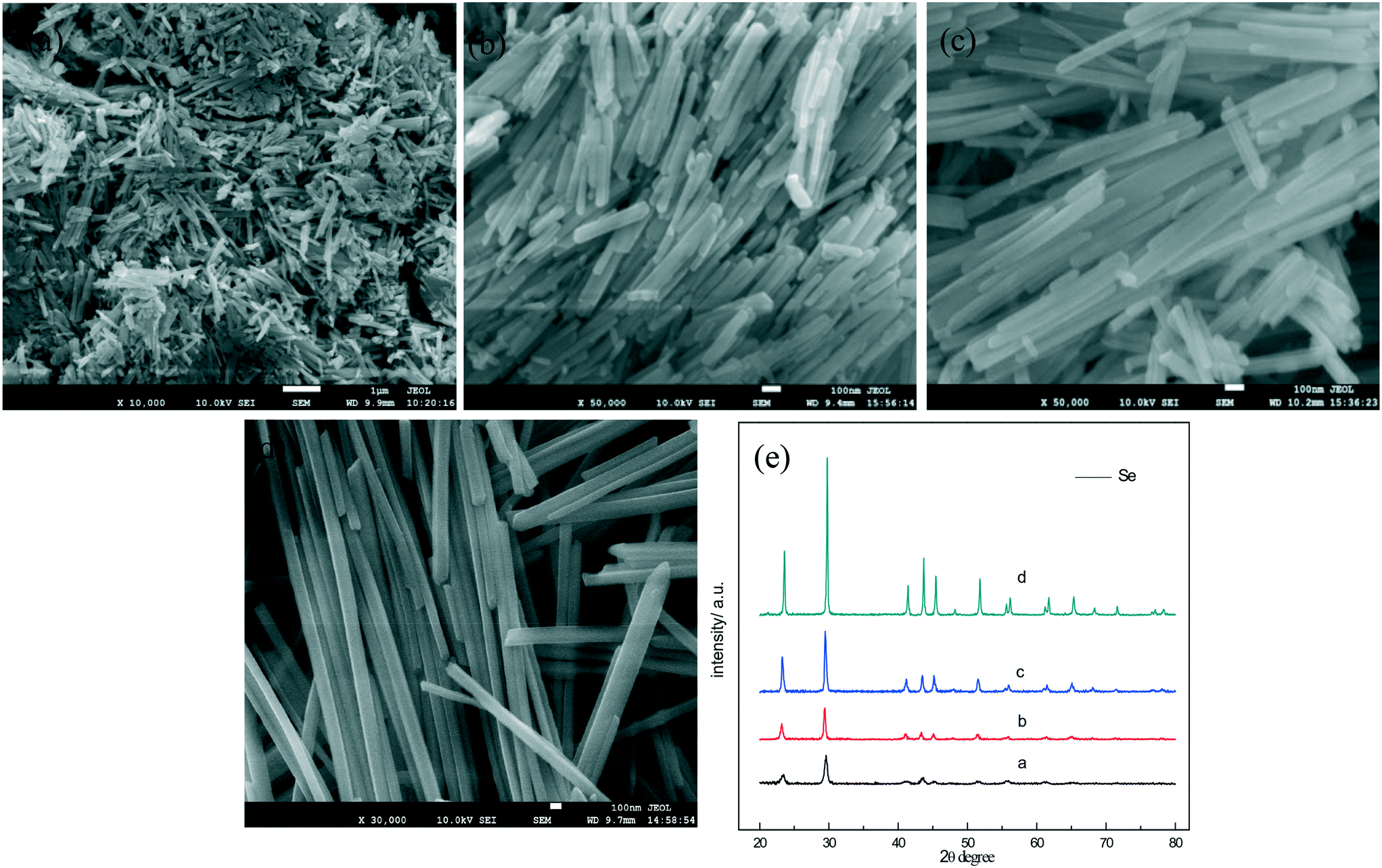
We also found that the concentration of a-Se in ethanol has an effect on the diameter of nanowires. In the present work, the selenium nanowires with different diameters were synthesized by controlling the concentration of amorphous selenium in ethanol; Fig. 7a–d are the SEM images of the t-Se nanowires with average diameters of 15, 26, 38 and 90 nm, respectively. When the concentration of a-Se increases to 1 g L−1, the t-Se nanowires become t-Se nanobelts (Fig. 7e and f). Fig. 7e indicates that the thickness, the width and the length of nanobelts is about 30 nm, 400 nm and several tens of micrometers, respectively. The dimensions were larger when the concentration of a-Se was increased to 2 g L−1 (Fig. 7f). The diameter of t-Se nanowires increases with increasing concentration of a-Se, finally forming nanobelts. This may be due to that there are a lot of t-Se seeds formed when the concentration of a-Se is high; these seeds most likely aggregated to increase the lateral dimensions of t-Se seeds (that is the diameter of the 1D structure), resulting in the formation of nanowires with larger diameter and even nanobelts. The corresponding XRD patterns of t-Se nanowires with different diameters and t-Se nanobelts are shown in Fig. 8.
 | ||
| Fig. 7 SEM images of t-Se nanowires (a–d) and nanobelts (e and f) prepared by dissolving (a) 0.005 g, (b) 0.01 g, (c) 0.02 g, (d) 0.05 g, (e) 0.1 g and (f) 0.2 g a-Se, respectively, into 100 mL ethanol. | ||
 | ||
| Fig. 8 XRD patterns of t-Se nanowires (a–d) and nanobelts (e and f) prepared by dissolving (a) 0.005 g, (b) 0.01 g, (c) 0.02 g, (d) 0.05 g, (e) 0.1 g and (f) 0.2 g a-Se, respectively, into 100 mL ethanol. | ||
3.3. Nanorods and nanotubes of t-Se
In the present experiment, we put the as-prepared a-Se into water for sonication, and selenium nanorods (Fig. 9a) were obtained after the color of the solution was changed from brick red to black-gray. It is seen from Fig. 9a that the morphology of the selenium nanorods is uniform, with an average diameter of 65 nm and a length of about 350 nm, which almost agrees with the particle size in the TEM image (Fig. 9b). The SAED pattern of t-Se nanorods indicates that these nanorods grew along the [001] direction, and the corresponding XRD pattern (Fig. 9c) shows that the sample is the trigonal phase of Se. It is worth noting that the sonication is a key factor in the preparation of t-Se nanorods, because if the a-Se solution is aged without sonication at room temperature for one month, no t-Se nanorods can be obtained and the brick-red solution of a-Se can be preserved all the time. | ||
| Fig. 9 (a) SEM image of t-Se nanorods prepared by adding 0.005 g a-Se into 20 mL water; (b) TEM image of t-Se nanorods, the insert shows the corresponding SAED pattern; (c) XRD pattern of t-Se nanorods. | ||
The effects of the solvents, dosage of a-Se, and PVP K30 on the growth of t-Se nanorods were investigated. When ethanol was used instead of water as the sonochemical solvent, t-Se nanorods were obtained after sonication for 10 min, but the diameter of the obtained t-Se nanorods was very uneven (Fig. 10a). This was most likely due to the much greater solubility of amorphous selenium in ethanol than in water,25,26 and a larger number of crystalline seeds were formed under sonication, which result in the formation of nanorods with irregular diameter.20 We also found that the length of nanorods increases with the increase in dosage of a-Se, which is shown in Fig. 10b–d, and the XRD patterns of these Se nanorods are presented in Fig. 10e.
 | ||
| Fig. 10 (a) SEM image of t-Se nanorods prepared using alcohol as the sonochemical solvent; SEM images of t-Se nanorods with different lengths prepared by adding (b) 0.01 g, (c) 0.03 g and (d) 0.05 g a-Se, respectively, into 20 mL water; (e) XRD patterns of t-Se nanorods. | ||
When PVP K30 was introduced together with a-Se into water, uniform t-Se nanotubes rather than nanorods can be obtained. The SEM images of t-Se nanotube shown in Fig. 11a and 10b indicate that the Se nanotubes were scattered around the center, with wall thicknesses about 50 nm, outer diameters in the range of 100–200 nm and lengths in the range 4–6 μm. Fig. 11c shows a typical TEM image, from which we can find that the dimension of t-Se nanotubes was almost consistent with the SEM result. The SAED pattern of t-Se nanotubes indicates that these t-Se nanotubes have predominantly grown along the [001] direction.21,27 The EDS spectrum (Fig. 11d) reveals Se and Cu as the major elements, and the Cu peaks originate from the TEM grid, which suggests that the samples are pure. All the diffraction peaks in the XRD pattern in Fig. 11e match well with the trigonal phase of Se.
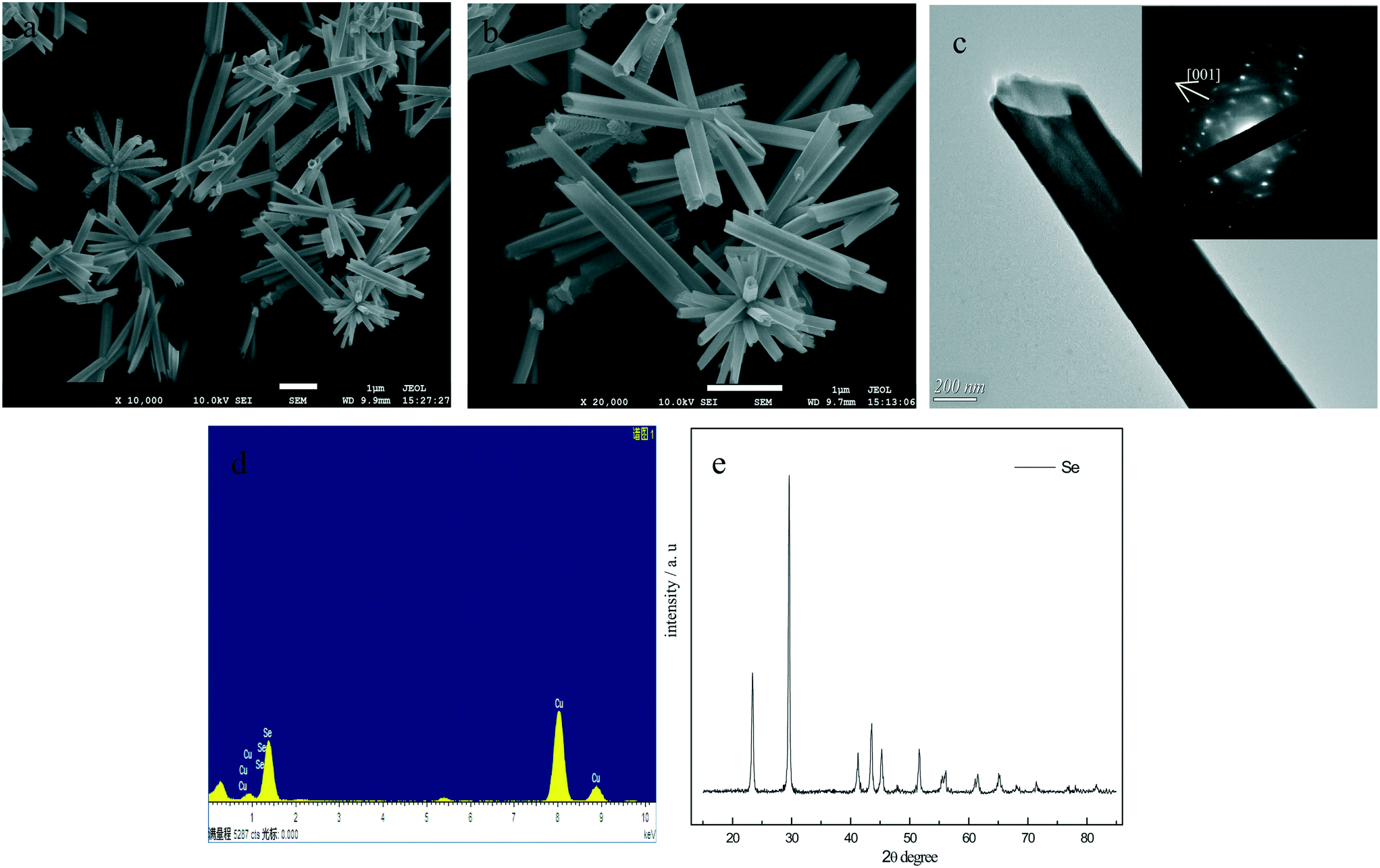 | ||
| Fig. 11 SEM images of t-Se nanotubes at a low magnification (a) and at a high magnification (b) prepared with 0.08 g a-Se together with 0.5 g PVP K30 added into 20 mL water; (c) TEM image of t-Se nanotubes, the insert shows the corresponding SAED pattern; (d) EDS spectrum of the t-Se nanotubes; (e) XRD pattern of t-Se nanotubes. | ||
Trigonal selenium nanorods and nanotubes can be obtained by the ultrasonication of a-Se nanospheres in water and with the addition of PVP for nanotubes, which indicates that the formation mechanism of t-Se nanotubes is the combination of the “solid–solution–solid” growth mechanism with the “surfactant-directed” growth mechanism. The possible formation mechanisms of Se nanotubes and Se nanorods are shown in Fig. 12 and 13. When PVP is introduced into the reaction system, a-Se nanoparticles are enveloped in PVP micelles, some of the Se atoms on the nanoparticle surface are dissociated into the micelles and the solution under ultrasonic irradiation (Fig. 12b), and then cylindrical t-Se seeds formed on the surfaces of a-Se nanoparticles (Fig. 12c). t-Se belongs to a hexagonal crystal system, which is similar to the crystal structure of Te nanotubes,27,28 when further addition of Se atoms to the surface of t-Se seeds would preferentially occur at the circumferential edges of cylindrical seeds because these sites had relatively higher free energies than other sites on the surface.28–30 As soon as the crystal growth began, mass transport to the growing regions would lead to undersaturation in the central portions of the growing faces, [001] planes, of each seed, resulting in the formation of the seeds of t-Se nanotubes (Fig. 12d), and the t-Se nanotubes were formed finally (Fig. 12e). As for the formation of t-Se nanorods, the formation of cylindrical t-Se seeds is similar to that of the nanotubes, and because there is no PVP effect, further addition of Se atoms to the surface of t-Se seeds would not only occur at the circumferential edges of the cylindrical seeds but also at the central portions of the growing faces ([001] planes) for each seed, which leads to the formation of nanorods (Fig. 13). In the experiment, we also found that longer t-Se nanorods were formed when the amount of a-Se nanoparticles was increased in water, which can be attributed to the sufficient Se atoms to extend the longitudinal dimension of the t-Se 1D nanostructure.
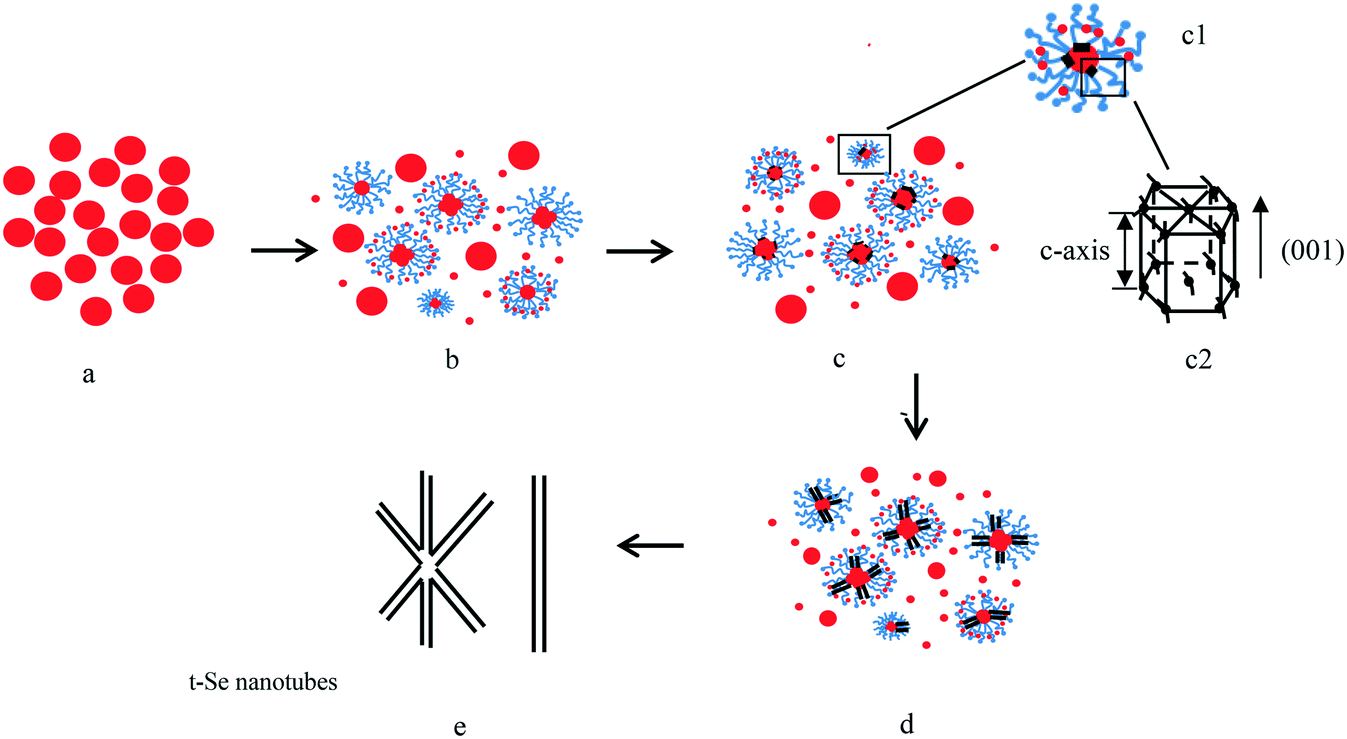 | ||
| Fig. 12 Schematic illustration of the formation of t-Se nanotubes. a) a-Se nanospheres; b) some a-Se colloids were surrounded by PVP to form micelles, meanwhile, a-Se nanospheres dissolved continuously; c) cylindrical t-Se seeds were formed on the surfaces of a-Se colloids; d) a-Se was continuously added into the circumferential edges of cylindrical seeds to form the seeds of t-Se nanotubes; e) all the a-Se colloids have been used up to form nanotubes. Note: the red, gray and blue colors represent a-Se, t-Se and PVP K30, respectively; c1 is a larger version of a micelle, c2 is the crystal structure of a t-Se seed. | ||
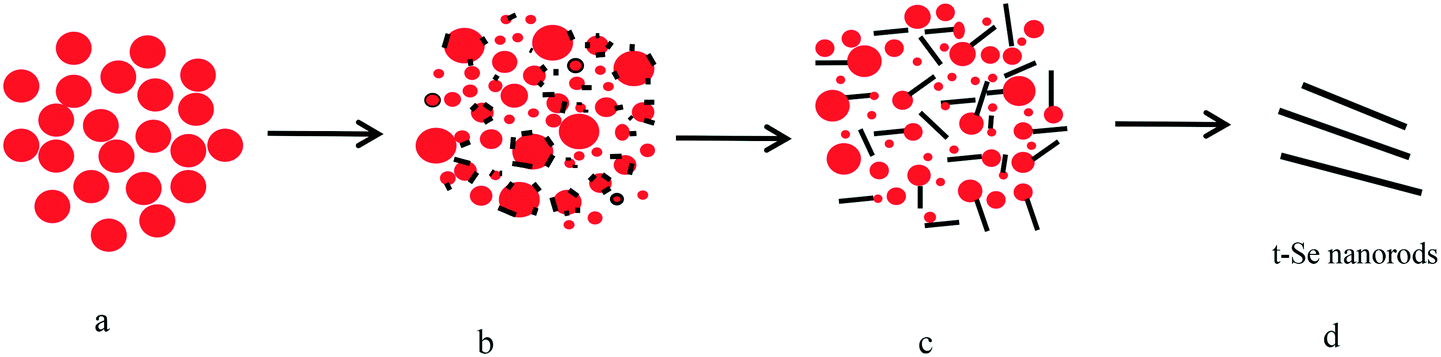 | ||
| Fig. 13 Schematic illustration of the formation of t-Se nanorods. a) a-Se nanospheres; b) the seeds of t-Se nanorods were formed on the surfaces of a-Se colloids, and a-Se nanospheres dissolved continuously. c) t-Se seeds grew continually along the c-axis at the expense of the gradually dissolved a-Se nanoparticles; d) all the a-Se colloids have been used up to form nanorods. | ||
3.4. Discussion on the formation mechanism of t-Se with different morphologies
In our experiment, we synthesized t-Se with different shapes via different a-Se aging processes. The schematic illustration of the formation of t-Se nanostructures with various morphologies is shown in Fig. 14. The conversion processes from a-Se nanospheres into t-Se 1D nanostructures comply with the “solid–solution–solid” formation mechanism, while the conversion from a-Se nanospheres into t-Se nanospheres complies with the mechanism of crystalline phase transformation. | ||
| Fig. 14 Schematic illustration of the formation of t-Se with various morphologies; I) ultrasonic irradiation; II) aging of a-Se in ethanol and in a dark environment; III) hydrothermal treatment. | ||
With respect to the growth mechanism of t-Se 1D nanomaterials, first, selenium is known to exist mainly in four allotropic forms including amorphous, trigonal, α-and β-monoclinic allotropes, of which the trigonal phase is the most stable crystal form and amorphous selenium can spontaneously transform into the trigonal phase. Second, the t-Se belongs to a hexagonal crystal system and it contains infinite helical Sen chain structures along the c-axis (Fig. 15), and hence, the t-Se could spontaneously develop into t-Se 1D nanostructures owing to its unique chains of atoms that favor anisotropic growth under appropriate conditions.31–35 Therefore, when the a-Se nanoparticles are present in solution, t-Se seeds would be generated initially under certain conditions, and then the t-Se can grow along the c-axis at the expense of the gradually dissolved a-Se nanoparticles owing to the intrinsically anisotropic structure of t-Se and eventually evolve into t-Se 1D nanostructures including nanowires, nanorods, nanobelts and nanotubes. In this case, the conversion processes from a-Se into t-Se 1D nanostructures comply with a typical “solid–solution–solid” formation mechanism, which is shown in Fig. 14 (routes I and II). In the formation mechanism, it was critical to choose a suitable solvent in which a-Se is soluble and t-Se is insoluble to allow for the growth of 1D t-Se nanostructures. In addition, the surfactant-directed growth mechanism plays an important role in the formation of t-Se nanotubes, which has been clarified above.
 | ||
| Fig. 15 The crystal structure of t-Se. | ||
The hydrothermal treatment can contribute to the amorphous-to-crystalline phase transition36 The t-Se nanospheres were synthesized by a hydrothermal method, and the schematic illustration of t-Se nanospheres is shown in Fig. 14 (route III). The t-Se nanoparticles prepared in our experiments almost have the same morphology and particle size as the a-Se precursor mainly due to the quick phase transformation under hydrothermal conditions.
In addition, t-Se nanorods and t-Se nanotubes can be prepared by the sonication process, while t-Se nanowires and t-Se nanobelts by aging the a-Se in ethanol. This is because a-Se is more soluble in ethanol than in water and t-Se 1D nanostructures grow faster in ethanol than in water under the same conditions, and therefore, the preparation of t-Se 1D nanostructures using water as the sonochemical solvent is generally achieved by ultrasonic irradiation.37,38 When using ethanol as the sonochemical solvent, nanorods with irregular diameters (Fig. 10a) were obtained instead of uniform nanowires. Therefore, it is better to prepare t-Se nanowires by aging the a-Se in ethanol naturally.
Up to now, in addition to utilizing the growth mechanism of “solid–solution–solid” to synthesize t-Se 1D nanostructures, t-Se 1D nanostructures can also be synthesized during recrystallization by dissolution of t-Se.38–40 In this case, the formation of t-Se 1D nanostructures follows the “t-Se dissolution and recrystallization” growth mechanism instead of “solid–solution–solid”, because t-Se is the most stable crystal form among its four common allotropic forms; the dissolution and recrystallization process of t-Se nanoparticles is very slow despite the aid of ultrasound irradiation,41 and hence, this growth mechanism is difficult to conduct actually unless at high temperatures or under alkaline conditions.41,42 In brief, the most economical and simplest method for synthesizing t-Se 1D nanostructures is that based on the growth mechanism of “solid–solution–solid”.
4. Conclusions
In summary, a precursor conversion method has been developed to prepare trigonal selenium (t-Se) with different morphologies (nanowires, nanorods, nanobelts, nanotubes and nanospheres). The amorphous selenium (a-Se) nanospheres as the precursor can be converted into t-Se nanomaterials of various sizes and shapes by changing the reaction concentration, solvent, sonication irradiation and surfactants. The conversion processes from a-Se nanospheres into t-Se nanostructures with various shapes (tubes, rods, belts and wires) comply with a “solid–solution–solid” formation mechanism, and the surfactant-directed growth mechanism plays an important role in the formation of t-Se nanotubes. This paper provides an environmentally friendly, easily controlled, efficient and low cost route for the synthesis of t-Se nanomaterials with various sizes and morphologies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful to the National Natural Science Foundation of China (No. 21373147 and No. 21573157) for providing financial support for the preparation and characterization of the test material.References
- L. B. Luo, J. S. Jie, Z. H. Chen, X. J. Zhang, X. Fan, G. D. Yuan, Z. B. He, W. F. Zhang, W. J. Zhang and S. T. Lee, J. Nanosci. Nanotechnol., 2009, 9, 6292–6298 CrossRef CAS PubMed
.
- Z. M. Liao, C. Hou, Q. Zhao, L. P. Liu and D. P. Yu, Appl. Phys. Lett., 2009, 95, 093104 CrossRef
- A. Qin, Z. Li, R. Yang, Y. Gu, Y. Liu and Z. L. Wang, Solid State Commun., 2008, 148, 145–147 CrossRef CAS
- P. Liu, Y. Ma, W. Cai, Z. Wang, L. Qi and D. Chen, Nanotechnology, 2007, 18, 7554–7560 Search PubMed
- L. Cheng, M. Shao and D. Chen, J. Mater. Sci., 2008, 19, 1209–1213 CAS
- S. Chaudhary, A. Umar and S. K. Mehta, Prog. Mater. Sci., 2016, 83, 270–329 CrossRef CAS
- C. Wang, Q. Hu, Y. Wei, D. Fang, W. Xu and Z. Luo, Ionics, 2017, 23, 3571–3579 CrossRef CAS
- Y. Zhong, M. Yang, X. Zhou and Z. Zhou, Mater. Horiz., 2015, 2, 553–566 RSC
- C. P. Yang, S. Xin, Y. X. Yin, H. Ye, J. Zhang and Y. G. Guo, Angew. Chem., Int. Ed., 2013, 52, 8363–8367 CrossRef CAS PubMed
- S. Jiang, Z. Zhang, Y. Lai, Y. Qu and X. W. Wang, J. Power Sources, 2014, 267, 394–404 CrossRef CAS
- X. Jiang, B. Mayers, Y. Wang, B. Cattle and Y. Xia, Chem. Phys. Lett., 2004, 385, 472–476 CrossRef CAS
- B. Gates, Y. Wu, Y. Yin, P. Yang and Y. Xia, J. Am. Chem. Soc., 2001, 123, 11500–11501 CrossRef CAS PubMed
- J. P. Ge, S. Xu, J. Zhuang, X. Wang, Q. Peng and Y. Li, Inorg. Chem., 2006, 45, 4922–4927 CrossRef CAS PubMed
- X. Jiang, B. Mayers, T. Herricks and Y. Xia, ChemInform, 2004, 15, 1740–1743 Search PubMed
- T. Huang and L. Qi, Nanotechnology, 2009, 20, 025606 CrossRef PubMed
- B. Zhang, W. Dai, X. Ye, F. Zuo and Y. Xie, Angew. Chem., 2010, 118, 2571–2574 Search PubMed
- A. Kumar, I. Sevonkae and D. V. Goia, J. Colloid Interface Sci., 2014, 416, 119–123 CrossRef CAS PubMed
- Z. Quan, P. Yang, C. Li, X. Zhang, J. Yang and J. Lin, Cryst. Growth Des., 2008, 8, 3834–3839 CAS
- K. Mondal and S. K. Srivastava, Mater. Chem. Phys., 2010, 124, 535–540 CrossRef CAS
- B. T. Mayers, K. Liu, A. David Sunderland and Y. Xia, Chem. Mater., 2003, 15, 3852–3858 CrossRef CAS
- B. Gates, Y. Yin and Y. Xia, J. Am. Chem. Soc., 2000, 122, 12582–12583 CrossRef CAS
- B. Wunderlich and H. C. Shu, J. Cryst. Growth, 1980, 48, 227–239 CrossRef CAS
- B. Gates, B. Mayers, B. Cattle and Y. Xia, Adv. Funct. Mater., 2002, 12, 219 CrossRef CAS
- H. Chen, D. W. Shin, J. G. Nam, K. W. Kwon and J. B. Yoo, Mater. Res. Bull., 2010, 45, 699–704 CrossRef CAS
- W. Zhu, H. Xu, W. Wang and J. Shi, Appl. Phys. A: Mater. Sci. Process., 2006, 83, 281–284 CrossRef CAS
- L. Liu, Q. Peng and Y. Li, Nano Res., 2008, 1, 403–411 CrossRef CAS
- Y. Ma, L. Qi, J. Ma and H. Cheng, Adv. Mater., 2004, 16, 1023–1026 CrossRef CAS
- B. Mayers and Y. Xia, Adv. Mater., 2002, 14, 279–282 CrossRef CAS
- G. C. Krueger and C. W. Miller, J. Chem. Phys., 1953, 21, 2018 CrossRef CAS
-
A. L. Barabasi and H. E. Stanley, Fractal concepts in surface growth, Cambridge University Press, Cambridge, 1995 Search PubMed
- F. Liao, X. Han, Y. Zhang, H. Chen and C. Xu, Mater. Lett., 2017, 214, 41–44 CrossRef
- G. P. Lindberg, Pressure and photo-induced modification of structural and chemical order in binary and elemental chalcogenide based materials, State University of New York at Buffalo, 2016 Search PubMed.
- X. Cao, Y. Xie, S. Zhang and F. Li, Adv. Mater., 2004, 16, 649–653 CrossRef CAS
- B. Gates, B. Mayers, B. Cattle and Y. Xia, Adv. Funct. Mater., 2010, 12, 219–227 CrossRef
- B. Gates, B. Mayers, A. Grossman and Y. Xia, Adv. Mater., 2002, 14, 1749–1752 CrossRef CAS
- P. Bettermann and F. Liebau, Contrib. Mineral. Petrol., 1975, 53, 25–36 CrossRef CAS
- Z. Chen, Y. Shen, A. Xie, J. Zhu, Z. Wu and F. Huang, Cryst. Growth Des., 2009, 9, 1327–1333 CAS
- X. Li, Y. Li, S. Li, W. Zhou, H. Chu, W. Chen, I. L. Li and Z. Tang, Cryst. Growth Des., 2005, 5, 911–916 CAS
- G. Xi, K. Xiong, Q. Zhao, R. Zhang, H. Zhang and Y. Qian, Cryst. Growth Des., 2006, 6, 577–582 CAS
- H. Zhang, D. Yang, Y. Ji, X. Ma, J. Xu and D. Que, J. Phys. Chem. B, 2004, 108, 1179–1182 CrossRef CAS
-
J. A. Dean, Lange's Handbook of Chemistry, ed. J. A. Dean, McGraw-Hill, New York, 1985 Search PubMed
- H. Zhang, D. Yang and X. Ma, Mater. Lett., 2009, 63, 1–4 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2018 |