
The crystalline sponge method: a solvent-based strategy to facilitate noncovalent ordered trapping of solid and liquid organic compounds†
Timothy R.
Ramadhar
 *a,
Shao-Liang
Zheng
b,
Yu-Sheng
Chen
c and
Jon
Clardy
*a
*a,
Shao-Liang
Zheng
b,
Yu-Sheng
Chen
c and
Jon
Clardy
*a
aDepartment of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, 240 Longwood Avenue, Boston, Massachusetts 02115, USA. E-mail: timothy_ramadhar@hms.harvard.edu; jon_clardy@hms.harvard.edu; Tel: +1 617 432 3801 Tel: +1 617 432 2845
bDepartment of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, Massachusetts 02138, USA
cChemMatCARS, Center for Advanced Radiation Sources, The University of Chicago c/o Advanced Photon Source, Argonne National Laboratory, 9700 South Cass Avenue, Argonne, Illinois 60439, USA
First published on 14th July 2017
Abstract
A strategy that leverages solvent effects to noncovalently trap solid and unstable liquid organic compounds within a crystalline sponge ({[(ZnI2)3(tris(4-pyridyl)-1,3,5-triazine)2]·x(CHCl3)}n) in a simple, mild, and efficient fashion for target molecule structure determination via X-ray diffraction is disclosed. Host–guest structures were obtained using third-generation synchrotron radiation, and new beamline hardware allowed rapid data collection in ∼5–24 minutes. This is 40–90% faster than previously reported crystalline sponge synchrotron datasets collected by us, and approximately a 150–720-fold decrease in time versus using a typical in-house diffractometer, effectively enabling the potential for high-throughput analysis. The new target molecule inclusion method using methyl tert-butyl ether (MTBE) solvent was demonstrated by trapping (E)-stilbene, vanillin, 4-(trifluoromethyl)phenyl azide, and (+)-artemisinin (an antimalarial drug). The potential of guests to maximize intermolecular interactions with the crystalline sponge framework at the expense of attenuating intramolecular interactions (e.g., π-conjugation) was observed for (E)-stilbene. Trapping of vanillin and (+)-artemisinin elicited single-crystal-to-single-crystal transformations where space group symmetry reduced from C2/c to P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and C2, respectively, and the absolute configuration of (+)-artemisinin was determined through anomalous dispersion.
and C2, respectively, and the absolute configuration of (+)-artemisinin was determined through anomalous dispersion.
Introduction
Single-crystal X-ray diffraction (SC-XRD) is one of the most powerful analytical techniques for small-molecule structure determination. One experiment can provide accurate information on atom types, bond lengths, relative stereochemistry, and absolute configuration. However, a major limitation for SC-XRD is the requirement of a single crystal, which traditionally precluded analysis of liquids and amorphous solids. A technique known as the “crystalline sponge method”1 reported by Fujita and co-workers circumvents this limitation by incorporating target molecules inside a crystalline metal organic framework (MOF), such as the commonly used crystal sponge {[(ZnI2)3(tris(4-pyridyl)-1,3,5-triazine)2]·x(solvent)}n (1) (Fig. 1),2 where target molecules become trapped in the MOF pores through intermolecular interactions with the host framework. The resulting host–guest complex can then be subjected to SC-XRD to determine the structure of the target molecule. Our work in this area has focused on improving the operational ease and reliability of the technique by reducing host preparation time, establishing crystallographic guidelines to ensure that chemically and physically-sensible models are constructed from high-quality data, and investigating the beneficial effects of changing the MOF terminal halide ligand.3,4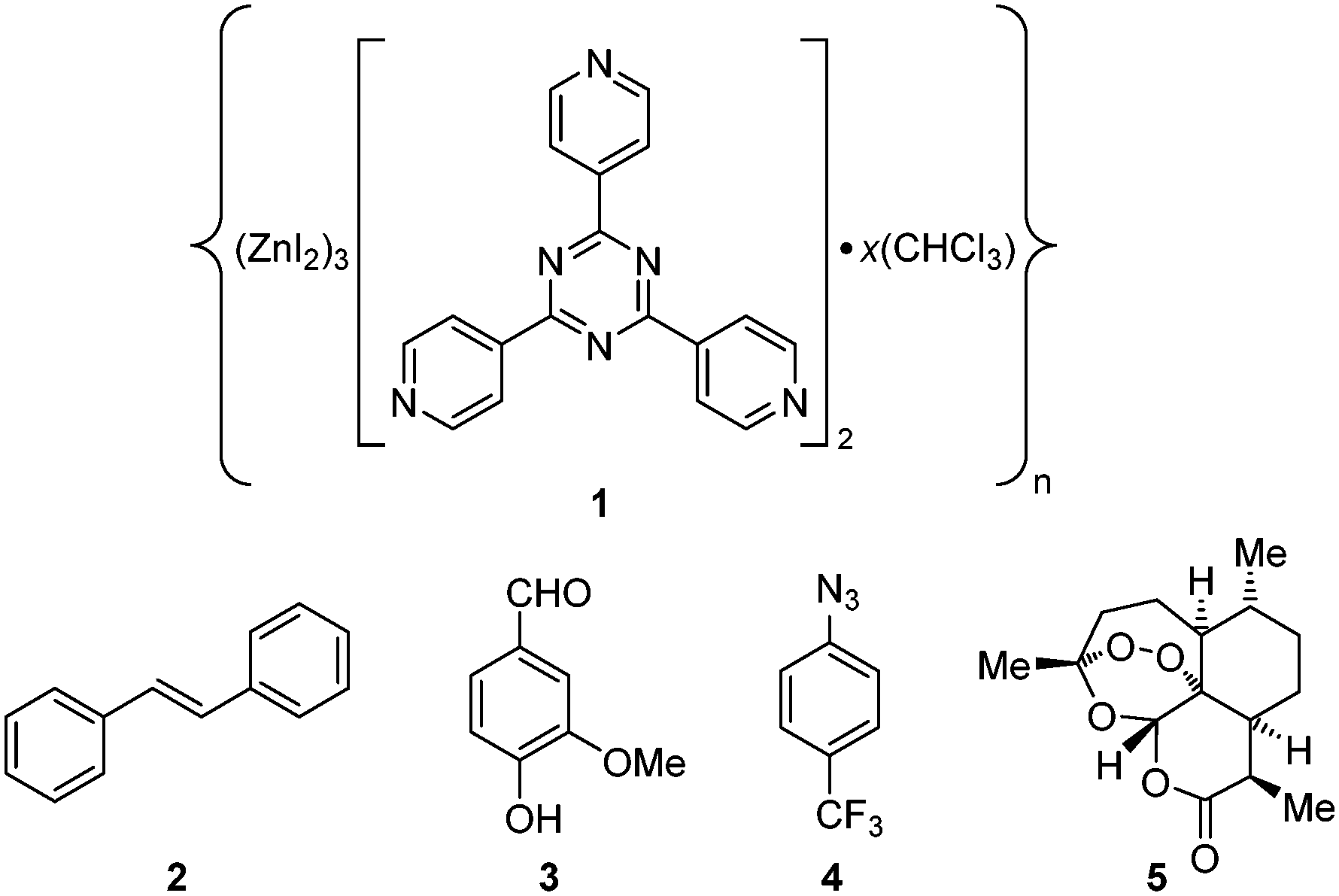 | ||
| Fig. 1 Structure of crystalline sponge and target molecules. | ||
The guest inclusion method that we previously disclosed involves soaking 1·CHCl3 with neat liquid compound.3 However, the ability to trap amorphous and crystalline solids in a rapid and operationally simple fashion in 1·CHCl3, which is easy and fast to synthesize, would expand the scope of compounds that can be analysed. Inclusion of crystalline solids is important if the native crystals exhibit poor shape (habit), diffract poorly, are prone to problematic twinning, or one wishes to determine absolute configuration of a molecule that does not contain heavy atoms and access to a diffractometer with a CuKα radiation source is unavailable. Furthermore, inclusion of solids allows for a unique opportunity to study reaction mechanisms for those compounds in situ by turning the crystalline sponges into “molecular flasks”.5 Fujita and co-workers have reported a guest inclusion protocol for solid and liquid compounds that uses cyclohexane with up to 10% CH2Cl2, CHCl3, or 1,2-dichloroethane to solubilize microgram quantities of a target compound.6,7 The resulting solution is added to 1·cyclohexane crystals and heated at 50 °C for multiple days to concentrate the target and facilitate guest penetration. While the procedure has been demonstrated by Fujita and co-workers, it is technically challenging to execute and typically requires multiple trials, which if conducted in parallel requires a significant amount of material. With extensive disorder and low occupancies it could also be possible to confuse residual solvent as target molecules if the target contains cyclohexyl rings, which may complicate structure refinement.3 In addition, cyclohexane (polarity index: 0, water insoluble)8 is not an optimal solvent for dissolving a wide array of compounds even if a small quantity of halogenated solvent is added. A recent report by Santarsiero and co-workers describing the fastest synthesis of 1·PhNO2 to date noted the use of n-Bu2O to solvate liquid diisopropylaniline for inclusion.9 While 1·PhNO2 exhibits a limited scope for guest trapping versus1·cyclohexane and 1·CHCl3 due to the affinity of PhNO2 for the host pores, the authors demonstrated that 1·PhNO2 can be converted to 1·cyclohexane and 1·CHCl3. However, like cyclohexane, the polarity of n-Bu2O (polarity index: 1.7, water insoluble)8 is not poised to dissolve a wide range of functionalized compounds. Our attempts to include targets in CHCl3 into 1·CHCl3 have failed likely due to higher solvent polarity (polarity index: 4.4, water solubility: 0.795 g/100 mL)8 that may favour target solvation and disfavour penetration and ordering within 1·CHCl3 (however, Carmalt and co-workers10 recently included naphthalene and anthracene saturated in CHCl3 into 1·CHCl3 over 1 week, presumably facilitated through strong guest π⋯π and CH⋯π interactions with the host).
To increase the scope of our procedure, we sought to find a solvent with a polarity that could strike a balance between compound solubilisation and allowing successful inclusion and ordering within 1·CHCl3. We believe that target solubilisation is important because maximizing target concentration can aid inclusion and identification as shown by Fujita and co-workers for nobiletin.7 We found that MTBE (polarity index: 2.5, water solubility: 5.1 g/100 mL),8 which is chemically tolerant of 1·CHCl3 unlike THF (where the latter presumably interacts with Zn2+ and displaces the tpt ligand), could strike that balance, and is contrary to a point raised by Fujita and co-workers stressing the necessity of using a poor solvent.7 Herein we report the simplest method to date for trapping solid and unstable liquid organic compounds in 1·CHCl3 (a “soak it and forget it” procedure), describe the use of third-generation synchrotron radiation with recent beamline hardware advances to drastically reduce the collection time and obtain high-quality data, demonstrate the method's effectiveness by trapping (E)-stilbene (2), vanillin (3), 4-(trifluoromethyl)phenyl azide (4) and (+)-artemisinin (5), and analyse the inclusion complexes.
Experimental
Guest inclusion procedure
The synthesis of crystalline sponge 1·CHCl3 was performed in 3 days as previously reported.3 Target compounds (2–5) were commercially obtained and solvated to maximum concentration up to 0.5 M in MTBE at room temperature (2: 0.14 M, 3: 0.5 M, 4: 0.5 M, 5: 0.07 M) (these are unoptimized; therefore it is possible to use lower concentrations), and 2 was also dissolved in CHCl3 at 0.5 M for comparison. For guest inclusion, multiple crystals of 1·CHCl3 were placed into a vial and the residual CHCl3 was removed through careful pipetting. Enough target compound in MTBE (or CHCl3) was added to submerge 1·CHCl3. At no point should the crystals ever become dry; thus, addition of the target compound in solvent should occur immediately after removal of residual CHCl3. The vial was capped, sealed with plastic paraffin film, and immediately shipped to the synchrotron. Soaking occurred at ambient temperature, and the soaking time was dependent on shipping and start of the synchrotron data collection shift. Compounds 2, 3, and 5 were soaked into 1·CHCl3 for 3 days, and 4 was performed for 2 days.Crystallographic procedure
Soaked crystalline sponges exhibiting prismatic habit were picked in NVH immersion oil at the beamline with polyimide loops. SC-XRD was performed using high-flux third-generation synchrotron radiation at the ChemMatCARS Sector 15 beamline (undulator insertion device) of the Advanced Photon Source in Argonne National Laboratory. For 1·2, 1·3, and 1·5, loops were mounted on a Huber three-circle goniometer with free κ with a Dectris PILATUS3 X CdTe 1 M shutterless‡ pixel array detector and a 100 K N2 cold stream generated by an Oxford Cryojet at experimental station 15-ID-D.11 An unattenuated beam with a wavelength of ∼0.41 Å (30 keV) was used with 0.2–1 s exposure times, and data were collected with multiple ϕ scans at 0.5° increments with ω and κ offsets using a 13 cm detector distance. For 1·4, the loop was mounted on a Bruker D8 three-circle fixed χ goniometer with an APEX II charge coupled device (CCD) detector and an Oxford Cryostream generated 100 K N2 cold stream at experimental station 15-ID-B,12 where a wavelength of ∼0.41 Å was used with a 0.2 s exposure time, and data was collected with multiple ϕ scans at 0.5° increments with ω offsets using a 7 cm detector distance. It is important to mention that the new experimental setup at station 15-ID-D allowed collection times of ∼5–24 minutes, which is at least a 40–90% reduction in time versus prior crystal sponge synchrotron data collected by us at 15-ID-B,3,4,12 and is roughly a 150–720-fold improvement in time versus collecting datasets using a typical in-house diffractometer with a sealed-tube anode source and an area detector (assuming a 2.5 day collection with 2 min per frame exposure time).11,13 Evaluation of the initial diffraction images and reciprocal lattice construction was performed to ascertain crystal quality. Data collection for the occasional crystal that appeared to be cracked was aborted. There was no need to diffract additional crystals of 1·2–1·5 after data collection for the first good-quality crystal since those crystals afforded publishable structures.Data were processed in the Bruker APEX3 software suite,14 where data integration was performed in SAINT,15 and SADABS16 was used for multi-scan absorption correction. The resolution cut-off for integration was set to 0.84 Å (sin(θ)/λ = 0.6), where I/σ ≥ 3.00 for 1·2 was 0.88 Å, 1·3 was 0.91 Å, 1·4 was 0.87 Å, and 1·5 was 0.97 Å. The data was solved using intrinsic phasing (SHELXTL XT-2014)17 and least-squares refinement on F2 was performed using SHELXL.18 The riding model was used for hydrogen addition, and the Brennan and Cowan anomalous scattering coefficients (f′, f′′, and cross-section in b/atom) were calculated for the irradiation wavelength and applied during refinement. Absolute structure parameters (Flack x, Hooft y, p3(true), p3(false), p3(racemic twin)) for 1·5 were calculated in PLATON/BIJVOETPAIR.19 Molecular visualization was performed in Olex2.20
Specific procedures and guidelines for crystalline sponge system refinement as previously reported were observed.3 As specifically rationalized in our guidelines and in congruence with our previous crystalline sponge structures,3,4 and as recommended by Spek for crystalline sponge systems,21 programs to treat residual density in solvent accessible voids for the reported and deposited structures were not used. For 1·2, there was a two-part disorder of 2 over an inversion center and one molecule of 2 involved in another two-part disorder was found on another inversion center. For these molecules it was necessary to manually insert the symmetry-related atoms and refine using negative SHELXL PART instructions. For all molecules of 2 in the asymmetric unit, it was necessary to use a SHELXL SAME directive with a smaller effective standard deviation (e.s.d.) for 1,2 and 1,3 distances (0.01 and 0.02, respectively) than the default values in order to rectify convergence issues (max. shift and max. shift/e.s.d. not converging to 0) and to deal with a level B checkCIF alert on phenyl C–C distances. For 1·3, it was necessary in some cases to change SHELXL AFIX 137 and AFIX 147 to AFIX 33 and AFIX 83, respectively, to deal with max. shift and max. shift/e.s.d. not converging to 0. For 1·5, disorder and the level of guest inclusion led to the normalized structure factor amplitudes exhibiting an <|E2 − 1|> of 0.899 (centric distribution) for space group determination; thus, it was necessary to force data processing in C2 versus C2/c, C2/m, and Cc. Electron density for what appears to be a heavily disordered molecule of 5 and MTBE was observed in the asymmetric unit; however, it could not be reasonably modelled and was left unmodelled in accordance with our reported guidelines.3
Results and discussion
Proof-of-concept for the facile trapping of solid organic compounds in 1·CHCl3 was established with (E)-stilbene (2). Compound 2 was dissolved in MTBE to 0.14 M and added to crystals of 1·CHCl3, upon which the crystals immediately turned a deep yellow colour. Soaking occurred at ambient temperature for 3 days before subjecting 1·2 to SC-XRD, and its structure was solved in centrosymmetric space group monoclinic C2/c where an R1 of 5.24% was afforded after refinement (Fig. 2a). The largest change in unit cell axis dimension upon guest inclusion was observed for the c-axis (1·2: c = 34.9049(14) Å versus1·CHCl3:§c = 31.081(3) Å). Three molecules of 2 were observed in the asymmetric unit where one was involved in a two-part disorder over an inversion centre (total occupancy: 50(1)%), one exhibited a two-part disorder where one disordered portion fell on an inversion centre (total occupancy: 73(1)%), and another molecule was observed at 81(1)% occupancy, where all were stabilized through host–guest intermolecular interactions.10,22 No molecules of MTBE or residual CHCl3 solvent were readily noticed. While the phenyl ring pairs for most guests have twist angles ranging between 2(2)–7(5)°, one disordered guest at 63.8(9)% occupancy exhibited an unusually large twist angle of 41(1)° (its disordered component exhibited a twist angle of 7(5)° and has a lower occupancy (9.5(5)%)) (Fig. 3a). The large twist angle for that guest mainly arises from its phenyl groups forging favourable contacts with the host and another proximal guest (Fig. 3b). This illustrates that guests can attenuate intramolecular interactions (π-conjugation in this case) in order to maximize favourable intermolecular contacts. When soaking was done with 2 solvated in CHCl3 to a higher concentration of 0.5 M, the crystals also immediately turned a deep yellow colour. However, 2 was not observed – only CHCl3 was found. Presumably while 2 penetrated 1·CHCl3 and elicited a colour change, the polarity of the CHCl3 solvent and/or its interactions with the framework precluded sufficient intermolecular stabilization of 2 with the host. Thus, 2 remained heavily disordered in the MOF voids and was not detectable by SC-XRD. In contrast, MTBE did not severely interfere with ordering of 2 and the latter was detectable at a lower concentration than when CHCl3 was used. It should be noted that a complex of 1 with 2 was previously reported by Fujita and co-workers, where 1·PhNO2 was soaked in (Z)-stilbene in cyclohexane (0.10 mmol/4 mL) and reacted using a Xe lamp (λex = 400–500 nm) for 155 h with 1 catalysing photoisomerization.23 The corresponding crystal structure of 1 with trapped 2 (R1: 12.21%, no disorder depicted) exhibits a similar arrangement of 2versus our reported 1·2 upon visually inspecting the packing models. However, large differences in inclusion method, data collection and refinement, presence of residual solvent in the voids (cyclohexane/PhNO2versus CHCl3/MTBE), and significant residual electron density in the former (max. peak: 9.1 e Å−3) makes drawing meaningful conclusions from detailed comparative analysis between the two complexes difficult.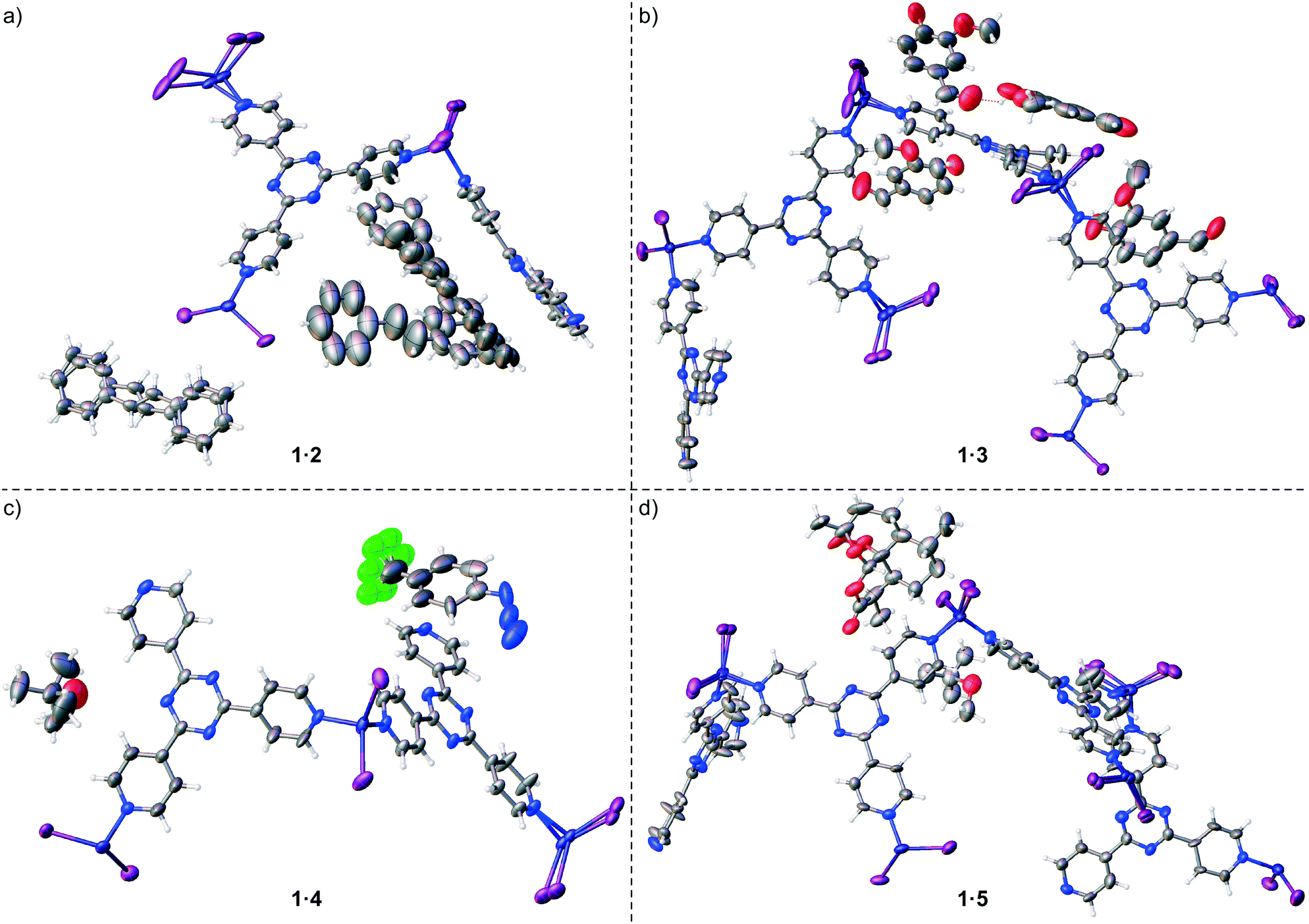 | ||
| Fig. 2 Asymmetric units for crystal structures of a) 1·2 (CCDC 1545812), b) 1·3 (CCDC 1545813), c) 1·4 (CCDC 1545814), and d) 1·5 (CCDC 1545815). Thermal ellipsoids are drawn at 50% probability. Guest and Zn/I host framework disorder is shown. | ||
 | ||
| Fig. 3 (a) Structure of a guest in 1·2 with an occupancy of 63.8(9)% and (b) its interactions with host 1 and another molecule of 2 at 81(1)% occupancy (packing model). Dashed green lines represent CH⋯π interactions and dashed black lines represent other van der Waals contacts. Ball-and-stick representation is shown, and other extraneous host/guest atoms have been hidden for clarity. | ||
The new procedure was applied to incorporate other solids and liquids into 1·CHCl3. Vanillin (3), a phenolic aldehyde that can be synthesized or isolated from processed Vanilla planifolia and Vanilla tahitensis seed pods and used as a flavouring/fragrance agent or in a popular TLC stain,24,25 was included into 1·CHCl3. Crystals of 1·CHCl3 were soaked in a 0.5 M solution of 3 in MTBE at ambient temperature for 3 days, where the crystals immediately developed a light yellow colour. Inclusion resulted in a single-crystal-to-single-crystal transformation affording a reduction in space group symmetry from C2/c to centrosymmetric triclinic P and different unit cell dimensions (1·3: a = 14.9223(8) Å, b = 18.9078(11) Å, c = 32.5910(18) Å, α = 102.7281(10)°, β = 91.7744(10)°, γ = 110.7963(9)° versus1·CHCl3:§a = 34.655(3) Å, b = 14.7307(14) Å, c = 31.081(3) Å, α = γ = 90°, β = 101.031(2)°) (Fig. 2b). Inspection of 1·3, exhibiting an R1 of 7.18%, revealed the presence of four molecules of 3 in the asymmetric unit with occupancies of 35(1)%, 41(1)%, 49(1)%, and 82(1)%. One molecule of 3 at 82(1)% occupancy had significant π⋯π stacking with a host pyridine ring (centroid⋯centroid: 3.801(8) Å), and its phenolic hydroxyl group hydrogen bonded to the aldehyde group in another molecule of 3 at 49(1)% occupancy to help anchor the latter within the sponge.
Trapping of 4-(trifluoromethyl)phenyl azide (4) in 1·CHCl3 through the new procedure was also successful. While organic azides are important precursors that participate in reactions such as Huisgen cycloadditions/click reactions, Staudinger ligations, Curtius rearrangements, Schmidt rearrangements, and [3,3]-sigmatropic shifts to afford useful molecules such as heterocycles and peptides, they are also high energy compounds that are potentially explosive.26 Azide 4 exists as an oil, however it is available for purchase as a 0.5 M solution in MTBE as a safer alternative for shipment and handling. We included 4 as a 0.5 M solution in MTBE into 1·CHCl3 upon soaking for 2 days at ambient temperature. The crystal structure of complex 1·4 was solved and refined in space group C2/c with an R1 of 5.76% (Fig. 2c). One molecule of 4 with a two-fold rotationally disordered CF3 group at roughly similar occupancy was observed in the asymmetric unit at 66(3)% occupancy, and one MTBE was present at 68(2)% occupancy. The electron density for the azide was unmistakably observed with an N–N–N angle of 170(3)°. Examination of the packing model revealed that the aryl-appended azide N forged an N⋯π contact with a pyridine (N⋯pyridine centroid: 3.86(2) Å), the central azide N formed a contact with an MTBE methoxy group (N⋯HC: 2.58(2) Å) and the terminal azide N formed contacts with the aforementioned MTBE methoxy group (N⋯HC: 2.80(3) Å) and a pyridyl C atom (N⋯C: 3.33(3) Å) (Fig. 4a). The MTBE also forged a CH⋯π interaction with the phenyl ring of 4via its tert-butyl methyl (CH⋯phenyl centroid: 2.939(9) Å), and the host framework established two additional CH⋯π interactions (CH⋯phenyl centroid: 3.06(1) Å, 3.29(1) Å). An alternating series of 4 and MTBE occurs in the channels where a F⋯HC contact between the CF3 of 4 and tert-butyl methyl of MTBE aids in its stabilization (minimum H⋯F: 2.76(5) Å) (Fig. 4b). The MTBE tert-butyl and methoxy groups can also be seen forging ancillary intermolecular contacts with the host. Overall, the success of this trapping experiment allows us to posit that liquid compounds that are unstable27 at high concentration can be included into 1·CHCl3 under mild dilute conditions using MTBE.
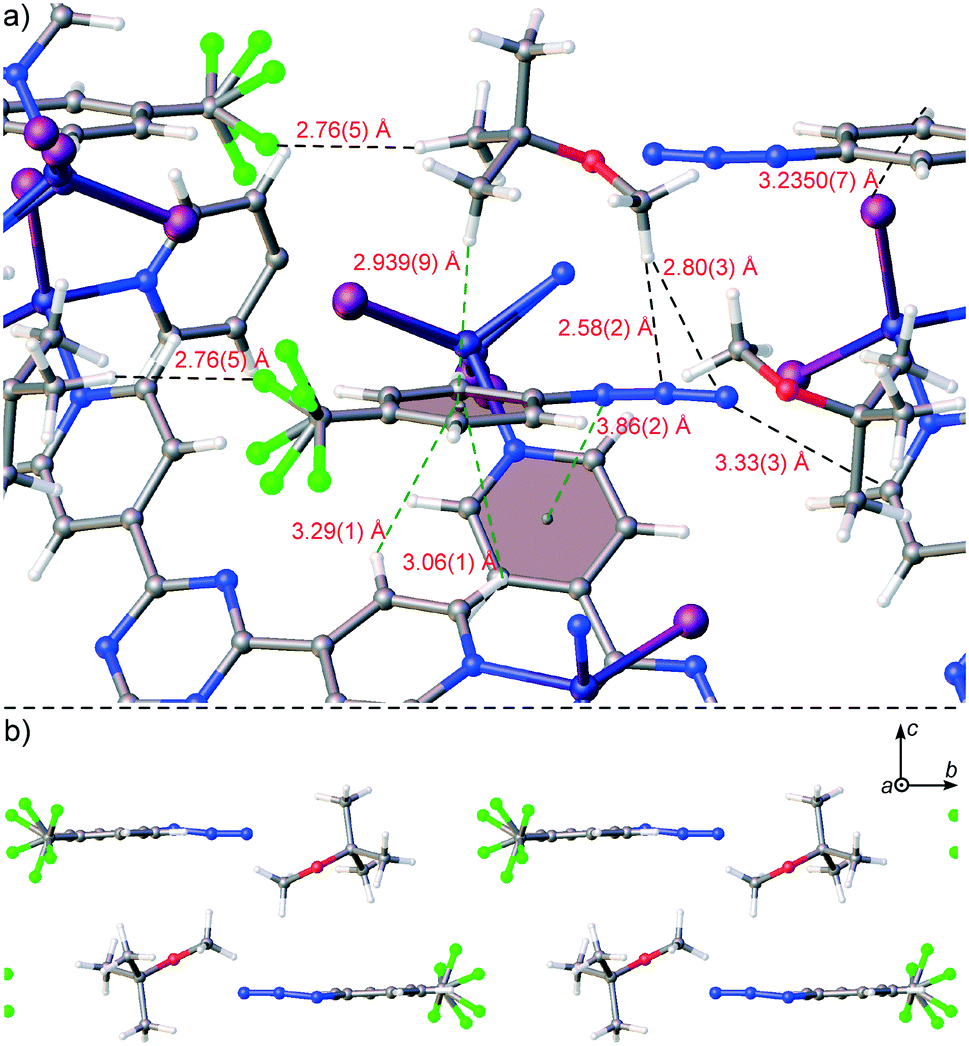 | ||
| Fig. 4 (a) Interaction of 4 with host framework 1 and MTBE in 1·4 (packing model) with extraneous atoms hidden for clarity and (b) an alternating series of 4 and MTBE within a channel of 1 with host framework 1 hidden. Ball-and-stick representation and CF3 disorder is shown. | ||
Finally, the new inclusion method was used to trap solid (+)-artemisinin (5) inside 1·CHCl3. Artemisinin (qinghaosu) is a tetracyclic sesquiterpene lactone endoperoxide natural product that is produced by sweet wormwood (Artemisia annua) and can be synthesized.28,29 It is used as a drug against Plasmodium falciparum malaria alongside other antimalarial agents (i.e., artemisinin-based combination therapies, ACT). Its use has saved millions of lives, and its discovery by Youyou Tu was part of the subject of the 2015 Nobel Prize in Physiology or Medicine.30 For the guest inclusion experiment, 5 was solubilized in MTBE to 0.07 M, added to 1·CHCl3, and soaking occurred at ambient temperature for 3 days. A single-crystal-to-single-crystal transformation occurred where the space group symmetry lowered from centrosymmetric C2/c to noncentrosymmetric monoclinic C2 – due to necessary destruction of inversion/c-glide symmetry upon chiral guest inclusion – to afford a chiral crystal system, and the largest change in unit cell axis was observed for the c-axis (1·5: c = 35.233(2) Å versus1·CHCl3:§c = 31.081(3) Å). An R1 of 7.00% was obtained after refinement of 1·5, and one molecule of 5 was clearly observed in the asymmetric unit with an occupancy of 93(1)% and one MTBE at 62(2)% occupancy (Fig. 2d). No anisotropic displacement parameter (ADP) restraints/constraints on 5 were required; only one soft C–C pairwise distance restraint was applied. In comparison to a single crystal structure of 5,31 the key peroxide O–O bond lengths and C–O–O–C torsion angles are similar (guest: 1.46(3) Å and 48(2)°, single crystal: 1.469(2) Å and 47.8(2)°). Some interesting interactions were observed in the packing model for 1·5 where the lactone carbonyl oxygen of 5 forged a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯π interaction with a host pyridine (O⋯pyridyl centroid: 3.22(2) Å) and the endocyclic oxygen in the oxepane ring that bears the endoperoxide forged an O⋯π interaction with a host triazine (O⋯triazyl centroid: 2.93(2) Å) (Fig. 5). The endocyclic lactone oxygen appears to form two O⋯π interactions (O⋯pyridyl centroid: 3.75(2) Å, O⋯triazyl centroid: 3.69(2) Å) and an oxepane methylene hydrogen formed a CH⋯π interaction with a pyridine (CH⋯pyridine centroid: 3.246(9) Å). These and other ancillary interactions, especially the first two described O⋯π contacts, play a significant role in anchoring 5 in the sponge. The Flack x and Hooft y absolute configuration parameters32,33 (0.18(3) and 0.15(2) respectively) are larger mainly due to unresolved disorder along with the use of a high-energy beam (30 keV, ∼0.41 Å) that diminishes the magnitude of the f′ and f′′ anomalous scattering coefficients and (to a lesser extent) “bad” reflections. The dextrorotatory absolute configuration of 5 was established through analysis of a three-hypothesis probabilistic model derived through Bayesian statistical analysis on Bijvoet differences using a Student's t-distribution (p3(true) = 1, p3(false) = 0, p3(racemic twin) = 3 × 10−38, Bijvoet pair coverage: 97%, correlation coefficient: 0.999) indicating that the absolute structure should not be inverted.33
O⋯π interaction with a host pyridine (O⋯pyridyl centroid: 3.22(2) Å) and the endocyclic oxygen in the oxepane ring that bears the endoperoxide forged an O⋯π interaction with a host triazine (O⋯triazyl centroid: 2.93(2) Å) (Fig. 5). The endocyclic lactone oxygen appears to form two O⋯π interactions (O⋯pyridyl centroid: 3.75(2) Å, O⋯triazyl centroid: 3.69(2) Å) and an oxepane methylene hydrogen formed a CH⋯π interaction with a pyridine (CH⋯pyridine centroid: 3.246(9) Å). These and other ancillary interactions, especially the first two described O⋯π contacts, play a significant role in anchoring 5 in the sponge. The Flack x and Hooft y absolute configuration parameters32,33 (0.18(3) and 0.15(2) respectively) are larger mainly due to unresolved disorder along with the use of a high-energy beam (30 keV, ∼0.41 Å) that diminishes the magnitude of the f′ and f′′ anomalous scattering coefficients and (to a lesser extent) “bad” reflections. The dextrorotatory absolute configuration of 5 was established through analysis of a three-hypothesis probabilistic model derived through Bayesian statistical analysis on Bijvoet differences using a Student's t-distribution (p3(true) = 1, p3(false) = 0, p3(racemic twin) = 3 × 10−38, Bijvoet pair coverage: 97%, correlation coefficient: 0.999) indicating that the absolute structure should not be inverted.33
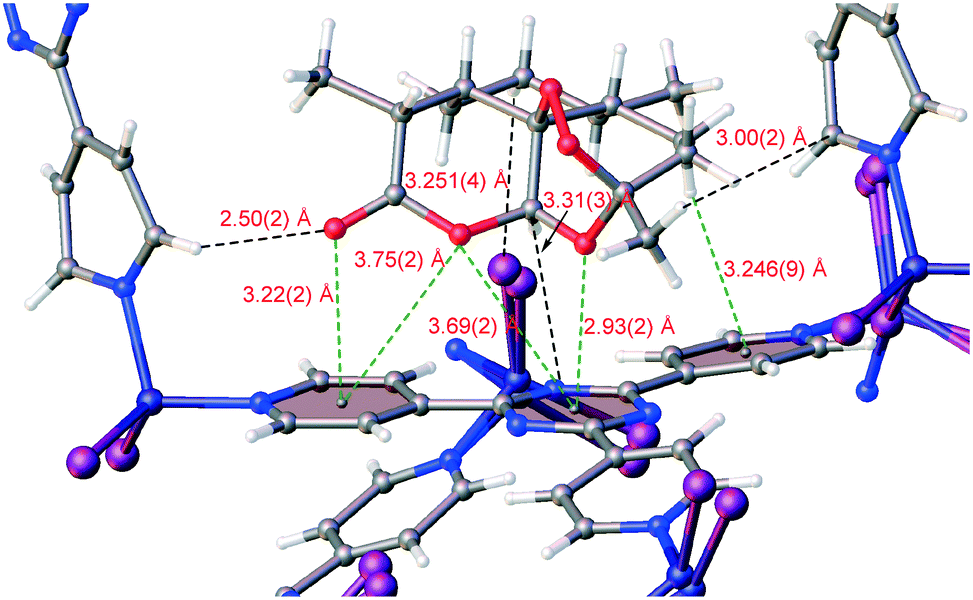 | ||
| Fig. 5 Interactions of 5 with host framework 1 in complex 1·5 as observed in the packing model. Ball-and-stick representation is shown and extraneous atoms have been hidden for clarity. | ||
Conclusions
Overall, we have reported a simple procedure to include solids and unstable organic liquid compounds in crystalline sponge 1·CHCl3 using MTBE. We demonstrated the new method by trapping both electron-rich and electron-deficient aromatic ((E)-stilbene, vanillin, 4-(trifluoromethyl)phenyl azide) and aliphatic ((+)-artemisinin) compounds. Analysis of the inclusion complexes revealed the intermolecular interactions involved in stabilizing guests within the pores of the crystalline sponge, and that guests can partially mitigate intramolecular interactions in order to maximize intermolecular contacts with the host and other guest molecules. The use of high-flux third-generation synchrotron radiation with a high dynamic range large-area shutterless detector reduced collection times to 5–24 minutes, which is an approximate 150–720-fold decrease in time versus using a typical in-house source. This increase in speed allows for high-throughput processing of various soaked crystalline sponges, and the use of a high-flux beam allows for high-quality data collection. It should be noted that the guest concentrations and soaking time have not been optimized; therefore, it is possible that these can be minimized. It is also very important to mention that not all compounds may enter the sponge through this method, or that they will enter and not be sufficiently stabilized through intermolecular interactions with the host framework to be detectable via SC-XRD.3 In cases where partial guest density is observed, we suggest trying other halide analogs of 1·CHCl3 as their contributions to the structure factors will be lower and thus may be easier to see guest density.4 It may also be advantageous to carefully attempt other known inclusion methods in especially problematic cases to determine whether a more successful result can be obtained. Finally, some compounds will be incompatible with 1 and cannot be analyzed through the current technique. Therefore the future of the crystalline sponge method must entail the development of new sponges to expand the scope of analyzable compounds.34 Nevertheless, the role of solvent in inclusion cannot be neglected, and we believe that the points made in this report will be applicable for use with other newly developed crystalline sponges.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful for financial support via the U.S. National Institutes of Health (U19-AI109673 and U19-TW009872 to J. C. and F32-GM108415 to T. R. R.). ChemMatCARS Sector 15 is principally supported by the Divisions of Chemistry (CHE) and Materials Research (DMR), National Science Foundation, under grant number NSF/CHE-1346572. Use of the PILATUS3 X CdTe 1M detector is supported by the National Science Foundation under the grant number NSF/DMR-1531283. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. We thank Dr. Su-Yin Grass Wang (University of Chicago) for beamline assistance.Notes and references
- Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 495, 461 CrossRef CAS PubMed
.
- K. Biradha and M. Fujita, Angew. Chem., Int. Ed., 2002, 41, 3392 CrossRef CAS PubMed
- T. R. Ramadhar, S.-L. Zheng, Y.-S. Chen and J. Clardy, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 46 CAS
- T. R. Ramadhar, S.-L. Zheng, Y.-S. Chen and J. Clardy, Chem. Commun., 2015, 51, 11252 RSC
- K. Ikemoto, Y. Inokuma, K. Rissanen and M. Fujita, J. Am. Chem. Soc., 2014, 136, 6892 CrossRef CAS PubMed
- Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai and M. Fujita, Nat. Protoc., 2014, 9, 246 CrossRef CAS PubMed
- M. Hoshino, A. Khutia, H. Xing, Y. Inokuma and M. Fujita, IUCrJ, 2016, 3, 139 CAS
-
(a) L. R. Snyder, J. Chromatogr. Sci., 1978, 16, 223 CrossRef CAS
- G. W. Waldhart, N. P. Mankad and B. D. Santarsiero, Org. Lett., 2016, 18, 6112 CrossRef CAS PubMed
- L. M. Hayes, N. J. Press, D. A. Tocher and C. J. Carmalt, Cryst. Growth Des., 2017, 17, 858 CAS
- Y.-S. Chen, H. Brewer, M. Meron and J. Viccaro, Acta Crystallogr., Sect. A: Found. Adv., 2014, 70, C1727 Search PubMed
- S.-Y. Wang, A. Stash and Y.-S. Chen, Manuscript in preparation.
- General example of the benefits of using a synchrotron versus an in-house source: B. J. Malbrecht, M. J. Campbell, Y.-S. Chen and S.-L. Zheng, J. Chem. Educ., 2016, 93, 1671 CrossRef CAS
-
Bruker, APEX3 (V2016.1-0), Bruker AXS Inc., Madison, Wisconsin, USA, 2016 Search PubMed
-
Bruker, SAINT (8.37A), Bruker AXS Inc., Madison, Wisconsin, USA, 2016 Search PubMed
-
Bruker, SADABS-2014/5, Bruker AXS Inc., Madison, Wisconsin, USA, 2014 Search PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 CrossRef PubMed
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed
- L. M. Hayes, C. E. Knapp, K. Y. Nathoo, N. J. Press, D. A. Tocher and C. J. Carmalt, Cryst. Growth Des., 2016, 16, 3465 CAS
- K. Ohara, Y. Inokuma and M. Fujita, Angew Chem., Int. Ed., 2010, 49, 5507 CrossRef CAS PubMed
- N. J. Gallage and B. L. Møller, Mol. Plant, 2015, 8, 40 CrossRef CAS PubMed
-
B. Spangenberg, C. F. Poole and W. Christel, Specific Staining Reactions, in Quantitative Thin-Layer Chromatography: A Practical Survey, Springer, Heidelberg, 2011, pp. 155–200 Search PubMed
- S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188 CrossRef PubMed
- S. Yoshioka, Y. Inokuma, V. Duplan, R. Dubey and M. Fujita, J. Am. Chem. Soc., 2016, 138, 10140 CrossRef CAS PubMed
- Y. Tu, Nat. Med., 2011, 17, 1217 CrossRef CAS PubMed
- M. A. Corsello and N. K. Garg, Nat. Prod. Rep., 2015, 32, 359 RSC
- E. Callaway and D. Cyranoski, Nature, 2015, 526, 174 CrossRef CAS PubMed
- J. M. Lisgarten, B. S. Potter, C. Bantuzeko and R. A. Palmer, J. Chem. Crystallogr., 1998, 28, 539 CrossRef CAS
- S. Parsons, H. D. Flack and T. Wagner, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 249 CAS
-
(a) R. W. W. Hooft, L. H. Straver and A. L. Spek, J. Appl. Crystallogr., 2008, 41, 96 CrossRef CAS PubMed
-
(a) E. Sanna, E. C. Escudero-Adán, A. Bauzá, P. Ballester, A. Frontera, C. Rotger and A. Costa, Chem. Sci., 2015, 6, 5466 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic data tables, solvent accessible void space analysis. CCDC 1545812–1545815. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ce00885f |
| ‡ Shutterless detectors do not require closure of the X-ray shutter for signal readout; it is done concurrently with irradiation. Thus, the readout step is eliminated making collection times shorter. |
| § See ref. 3 and CCDC 1007932 for 1·CHCl3. |
| This journal is © The Royal Society of Chemistry 2017 |