
Unravelling chromism in metal–organic frameworks
Gift
Mehlana
 *a and
Susan A.
Bourne
*b
*a and
Susan A.
Bourne
*b
aDepartment of Chemical Technology, Faculty of Science and Technology, Midlands State University, Private Bag 9055 Senga Road, Gweru, Zimbabwe
bCentre for Supramolecular Chemistry Research, Department of Chemistry, University of Cape Town, Rondebosch 7701, South Africa. E-mail: susan.bourne@uct.ac.za
First published on 22nd June 2017
Abstract
This paper examines chromic metal–organic frameworks, an important class of materials owing to their applications in several technological fields. These materials change their absorption spectra upon application of an external stimuli such as heat, pressure, light etc. This review explores recent advancements in photochromic, thermochromic, solvatochromic, and piezochromic metal–organic frameworks (MOFs). Emphasis is directed towards the design of chromic MOFs as well as the mechanisms associated with the colour change upon application of an external stimulus. Compared to traditional organic and inorganic materials, MOFs show exceptional chromic behavior as they can be fine-tuned to tailor the desired properties.
 Gift Mehlana | Gift Mehlana obtained his PhD from the University of Cape Town in 2014 and is currently a lecturer at the Midlands State University in Zimbabwe. His doctoral thesis looked at mechanistic aspects of dynamic metal organic frameworks for use in sensing and capturing of small molecules. His current research interests include the synthesis and characterisation of metal organic frameworks and coordination polymers for applications in catalysis and sensing. |
 Susan Bourne | Susan Bourne is Professor of Physical Chemistry at the University of Cape Town, South Africa. She received her BSc(Hons) degree in 1987 and her PhD in 1991, both at the University of Cape Town. After postdoctoral experience at Texas Christian University, USA, she took up a lecturing post at the University of Cape Town. Her research interests focus on crystal engineering of supramolecular systems with a particular interest in their use for chemical sensing and storage of unstable compounds. |
1. Introduction
Metal–organic frameworks (MOFs) as a class of crystalline materials constructed from metal ions and organic linkers (organic ligands with divergent chelating groups) have recently attracted a great deal of attention.1–4 Studies have shown that MOFs have many potential applications such as gas storage,5 sensing,6 separation,7 drug delivery8 and catalysis.9 Stimuli responsive MOFs which exhibit changes to temperature changes, solvent molecules, light or pressure have gained increasing attention recently for their potential applications in many areas.10 These materials can lead to greater control over guest selectivity as well as extending the range of practical applications to include sensors and actuators.11 Articles reporting on MOFs as sensing materials have undergone an exponential growth (Fig. 1).12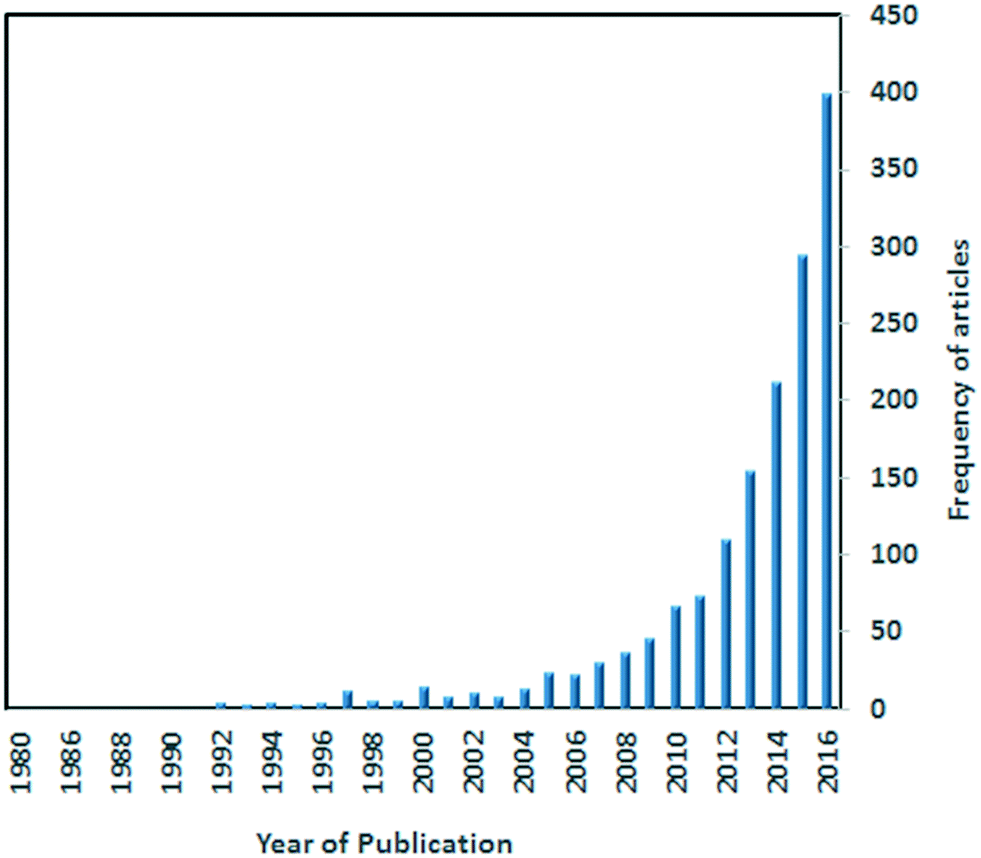 | ||
| Fig. 1 A Scifinder search showing the exponential increase in the number of published articles on MOFs as sensors. | ||
Recently, chromism (visible, often reversible, changes in colour) has emerged as a property of great potential for practical MOF-based devices. Colour changes can be caused by photo-irradiation, heat, compression, or solvent effects; the phenomena are correspondingly called photochromism, thermochromism, piezochromism, and solvatochromism. A commonly used strategy to construct these smart MOFs is to incorporate the stimuli responsive units as linkers in the framework. However, certain challenges have to be addressed for the successful design of these smart materials. For example, interpenetration in MOFs may prevent cis/trans isomerization reactions (rendering the MOF inactive towards light) or could greatly reduce the pore sizes (thus rendering a MOF non-porous and inactive to solvatochromic effects). While interpenetration can therefore be regarded as a hindrance in designing photochromic or solvatochromic MOFs, this phenomenon has advantages when designing redox active systems.
A number of photochromic MOFs have been reported which are based on loading or functionalization of azobenzene derivatives inside the pores.8,12–17 However, for photochromism to be realized in host guest inclusion compounds, it is imperative that the MOF has large pores to allow for the cis/trans isomerisation of the azobenzene guest material. Furthermore, the MOF framework must be chemically stable so as to withstand the conformational rearrangement of the chromic guest material within the pores. It is possible that during the isomerization process, the host framework maybe transformed into another form capable of rendering the reaction reversible.
There is precedence in literature that both photochromism and thermochromism can exist in a single MOF.18 While the two phenomena are associated with different mechanisms in the majority of reported cases, recent studies have revealed that thermochromism may also be ascribed to the generation of radicals, a mechanism that is common in photochromic systems.19 For solvatochromic systems the change in the absorption spectra is normally attributed to the interactions of the guest molecules with the host framework. Changes in the geometry of the metal centre have an impact on the colour change but such processes are not desirable as they often lead to distortion of the framework. While solvatochromism is reversible upon uptake of organic solvents/vapours, adsorption of water leads to an irreversible colour change associated with poisoning of the framework.20 It has been demonstrated in several studies that MOFs can be easily functionalized to achieve the desired properties.21,22 This implies that it is possible to design MOFs that can respond to specific solvent molecules to give a desirable colour change.
In this contribution, we describe the progress made over the past 5 to 10 years in developing MOFs that exhibit chromic effects which can be detected by the naked eye. Excellent reviews on luminescence in MOFs have been published23,24 so this phenomenon is not covered here. We focus on recent reports of chromic MOFs and their potential applications. Colour changes that are readily visible render MOFs as suitable sensors for changes in temperature, light, pressure, and the presence of volatile organic compounds. We have structured this review to consider photo-, solvato-, thermo-, and piezochromic MOFs in turn. In each section the focus is on the design principles and the mechanisms associated with chromic effects in MOFs. This in turn will assist in the engineering of materials that can respond to external stimuli for practical applications.
2. Photochromism
Photochromic materials are able to undergo reversible change in their molecular and electronic structure upon application of light.25 Comprehensive reviews of organic–inorganic hybrid photochromic materials have recently been published.26,27 These materials find applications in smart windows, data storage, photo catalysis as well as solar energy conversion.26 Photochromic properties of organic and inorganic compounds have been well explored. Inorganic photochromic compounds have considerable thermal stability, high strength and diverse coordination chemistry contrary to organic materials which have the advantage that they can easily be modified. In organic systems, photochromism is as a result of a photo-induced structural transformation between two chemical isomers that have different absorption spectra. This is normally accompanied by a chemical bond rearrangement which leads to structural and electronic changes of the molecules (Fig. 2).28–30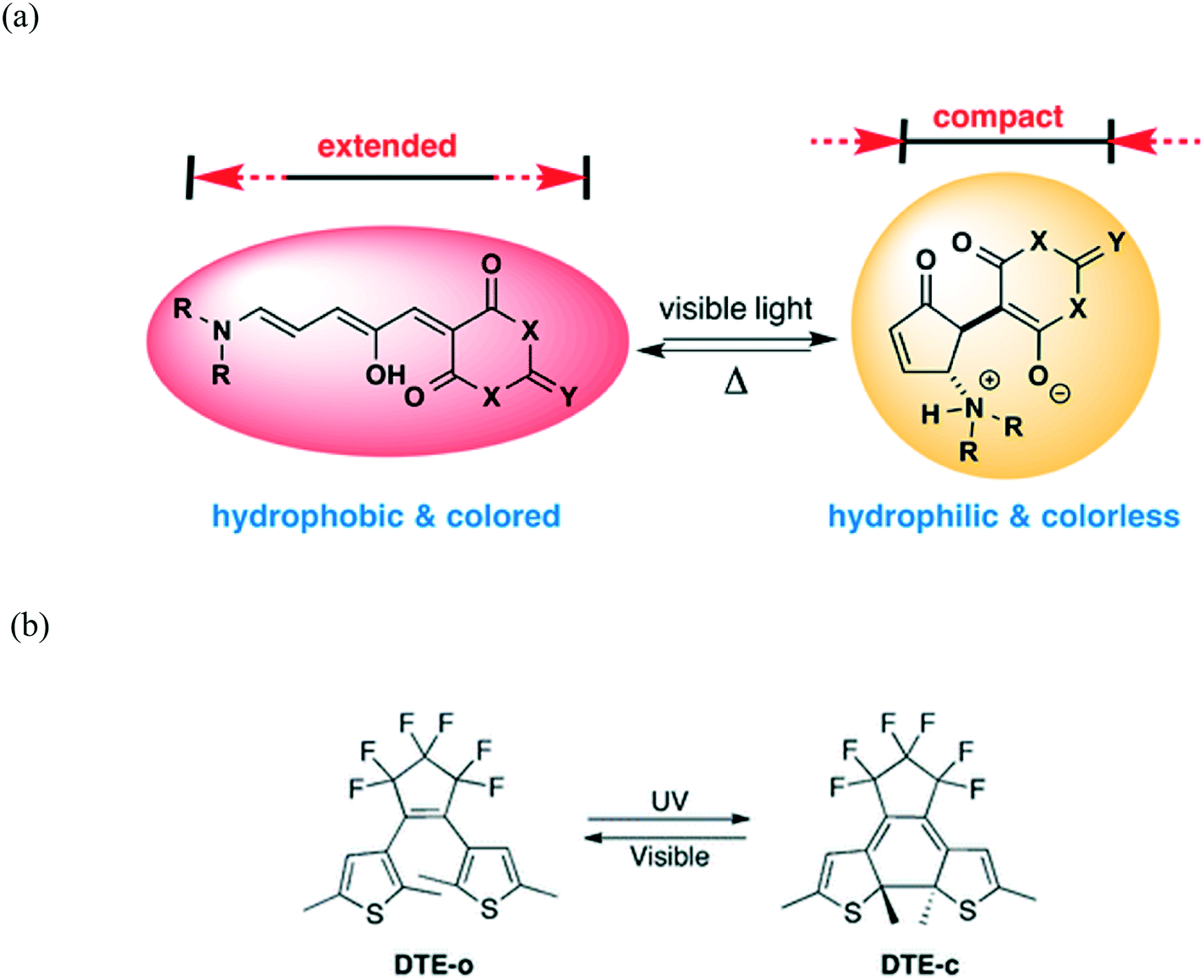 | ||
| Fig. 2 (a) An organic compound in the conjugated, coloured, and hydrophobic form (left) converts to a ring-closed, colourless zwitterionic structure (right) on irradiation with visible light. Reprinted with permission from ref. 28. Copyright (2014) American Chemical Society. (b) Photochromic reaction and molecular structure of 1,2-bis(2,5-dimethyl-thien-3-yl)perfluorocyclopentene (DTE) in the open (DTE-o, colourless) and closed (DTE-c, red) forms. Reproduced from ref. 30 with permission from the Royal Society of Chemistry. | ||
Molecules such as azobenzene, spiropyran, furylfulgide, and diarylethene, and their polymers can be photoswitched between two different colour states.14,29 It should be noted that the coloration is an energetically uphill process, hence molecules return to their original colour when the light is removed. A diarylethene family of compounds can switch from colourless to red under UV exposure via a structural transformation from an open-ring to a closed-ring isomer (Fig. 2b).30 Compared to other photochromic organic compounds, diarylethene has a thermally stable coloured state, which cannot be bleached in the dark at room temperature. Thus, the combination of organic and inorganic units in a single material can preserve or even improve the respective features of the inorganic and organic components.
2.1 Design aspects of photochromic MOFs
MOFs present a good platform for the design of photochromic materials owing to their porosity and dynamic nature. There are several approaches that can be used to design photochromic MOFs. The most common method is the use of a photochromic linker such as 1,2-bis(2-methyl-5-(pyridin-4-yl)thiophen-3-yl)cyclopent-1-ene (MTE) that is able to undergo some structural change upon light irradiation. Alternatively, bipyridinium carboxylate linkers can be used to construct photochromic MOFs since they are able to generate viologen radicals when irradiated with light.31 Viologens are relatively active and unstable in the presence of molecular oxygen and other radical scavengers. In light of this, it is of paramount importance to develop effective strategies to stabilize the photogenerated radicals.Naphthalene diimides (NDIs) are an attractive class of electron-deficient dyes which are able undergo a reversible one-electron reduction to form stable radical anions in the presence of electron donors upon irradiation. These compounds can used as linkers in the synthesis of photochromic MOFs. Polyoxometalates (POMs) form a unique class of multi-functional anionic metal oxygen clusters which can be introduced into the pores of NDI-based MOFs since they exhibit good redox and photo-activity, and can act as electron donors in photo-induced electron transfer processes. POMs mainly serve as photosensitizers to reinforce NDI radical generation, thereby increasing the rate of the photochromic process.32,33
Due to their inherent porosity, MOFs can also be loaded with photoactive materials such viologen cations which are known to undergo some electron transfer upon irradiation as well as molecules that are known to change colour as a result of cis–trans isomerisation process. The success of this method entirely depends on the size of the pores within the MOF. For example loading a MOF with azobenzene will require that there be enough space to allow for the cis–trans conformational rearrangement of the guest molecules. The disadvantage of this method is that it greatly reduces the pore size which may limit the practical applications of these materials. Furthermore it is difficult to control the orientation of the photoactive guest molecules within the pores of the MOFs. Thermal treatment and liquid-phase epitaxial layer-by-layer growth methods have been applied successfully to introduce guest molecules into the MOFs (Fig. 3).34
 | ||
| Fig. 3 Top; Reversible cis–trans isomerisation of azobenzene. Bottom; In situ growth of trans-azobenzene encapsulated HKUST-1 thin film using liquid-phase epitaxial layer-by-layer method. Reprinted with permission from ref. 33. Copyright (2016), American Chemical Society. | ||
Photochromic MOFs without photochromic organic linkers can be engineered by making use of the photoinduced bistable systems based on different electron-transfer mechanisms. The photochromic mechanisms include metal-centered electron transition (MC), ligand-to-metal charge transfer (LMCT), metal-to-metal CT (MMCT), intraligand CT (ILCT), and ligand-to-ligand CT (LLCT). Electron-transfer photochromic MOFs are electronically labile with their two or more electronic states lying close in energy, which leads to significant vibronic interactions and an appreciable sensitivity to external photo perturbations.
Recent studies have demonstrated that functionalizing the linkers with the photoswitchable functionality as a side-group seems a more reliable and successful approach.35,36 This design strategy can be achieved via post synthetic modification. It is of paramount importance to ensure that linker fits in the pores and also that its light-triggered isomerization mechanism can occur and that the resulting isomeric form can be accommodated within the resulting lattice. Wang et al. noted that small differences in the framework dimensions have been shown to suppress the photoresponse of Cu-containing surface-attached metal–organic frameworks (SURMOF) due to the lack of a free trajectory for the photoisomerization mechanism of the azobenzene pending groups (Fig. 4).37
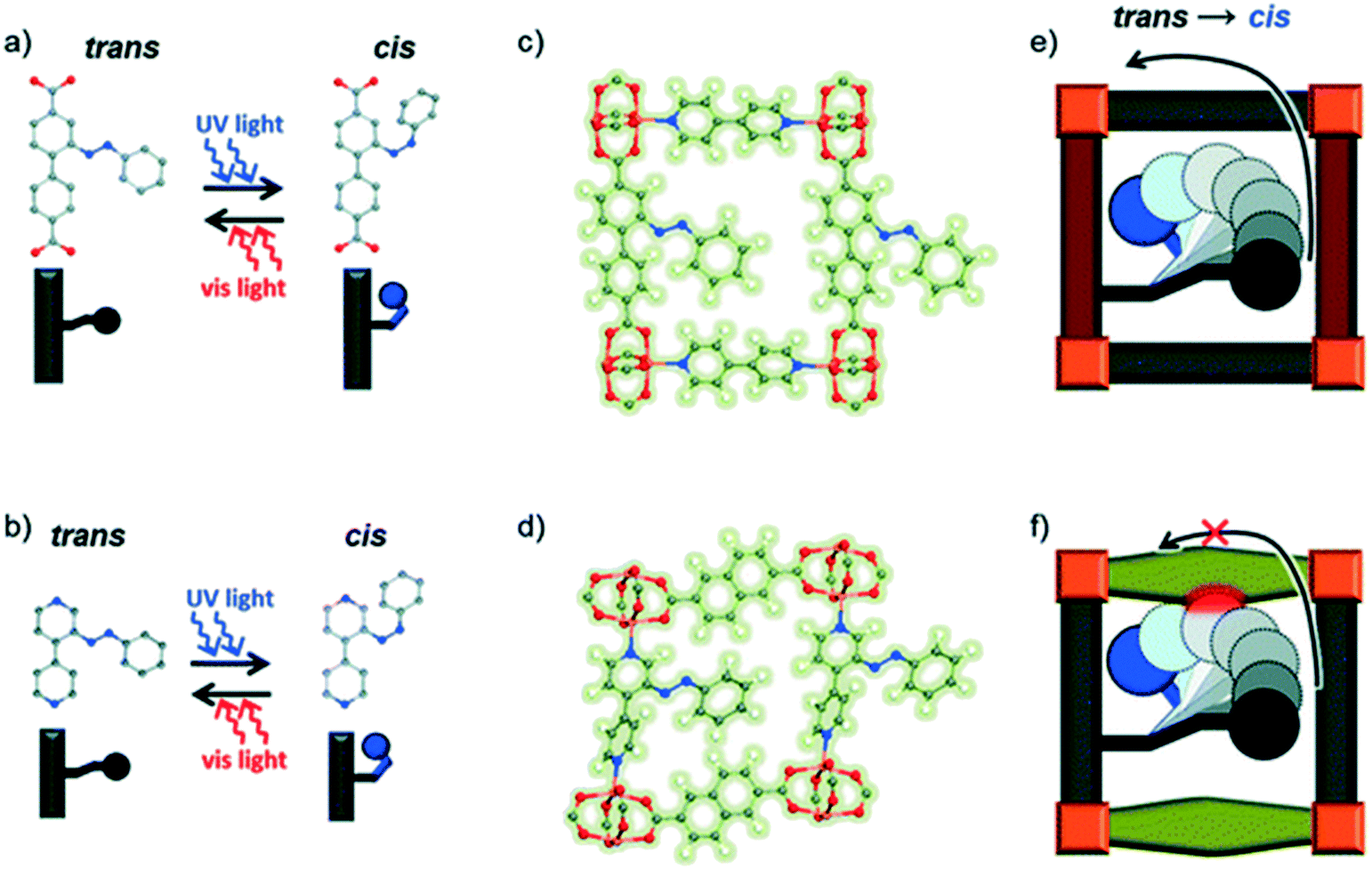 | ||
| Fig. 4 An illustration of the effect of linker length on the cis–trans isomerization of azobenzene in SURMOFs. (a) and (b): Linkers in the planar trans state can be switched to the cis state by UV light. (c) and (d) show side views of the relevant MOFs with azobenzene side groups in the trans state. Photodimerization is enabled in the MOF containing the longer linker (e), while that containing the shorter linker (f) is sterically hindered. Reproduced from ref. 37 with permission from the PCCP owner societies. | ||
Table 1 gives a general overview of the properties of the photochromic MOFs discussed in this review. Of particular interest to note is the response time of these MOFs upon irradiation with light, as well as the stability of the photo product. An understanding of these two properties will allow for the design of reversible fast responsive materials which can be used as sensors
| MOF | Linkers | Stimuli | Response time | Stability of photo product | Ref. |
|---|---|---|---|---|---|
| DMOF-1@DTE | Terephthalate (pBDC) and triethylenediamine (TEDA) | UV | Immediate | Stable in the dark for 1 month | 30 |
| [Cd3(BTC)2]n2− | 1,2,4,5-Benzenetetracarboxylate (BTC) | 300 W xenon lamp | 1 min | Stable in the dark for 2 months | 38 |
| [PV][Zn3(m-BDC)4]·H2O | 1,3-Benzenedicarboxylate (mBDC) | 150 W xenon lamp | 50 s | Stable for 12 h in the dark | 39 |
| MOF-808 | 1,3,5-Benzenetricarboxylate (TMA) | UV | — | — | 40 |
| [Zn(NDI-ATZ)(DMF)2]n | Naphthalenediide related tetrazole (NDI-ATZ) | Sunlight | 15 min | Stable in the dark for 1 h | 41 |
| PC-PCN | 1,2-Bis(2-methyl-5-(pyridin-4-yl)thiophen-3-yl)cyclopent-1-ene (BPDTE) | UV | 10 min | 1 h under visible light | 42 |
| UBMOF-1 | 9,10-Bis(2,5-dimethylthiophen-3-yl)-phenanthrene-2,7-dicarboxylate (PCA) | UV | Not given | Not reversible | 17 |
| [Zn(ipbp)(H2O)]·NO3·H2O | 1-(3,5-Dicarboxyphenyl)-4,4′-pyridinium bromide (H2ipbpBr) | UV/visible light | 5 min | 1 day | 43 |
| [Zn(ipbp)(H2O2)]·NO3·H2O | 1-(3,5-Dicarboxyphenyl)-4,4′-pyridinium bromide (H2ipbpBr) | UV/visible light | 10 min | Stable in the dark for several days | 43 |
| [Co(DCBPY)(N3)4] | N,N′-Bis(3,5-dicarboxylatobenzyl)-4,4′-bipyridinium (DCPBY) | Xenon lamp | Not given | Stable in air for several days | 19 |
| [Cd(CPBPY)(pBDC)(H2O)]n | N-(3-Carboxyphenyl)-4,4′-bipyridinium (CPBPY) and terephthalate (pBDC) | UV/xenon lamp | 1 min | Stable for months in the dark | 46 |
| Tb-MOF | 1-(3,5-Dicarboxybenzyl)-4,4′-bipyridinium chloride (H2ipbpCl) | 300 W xenon lamp | Not given | Decolours in 1 day | 47 |
| [Zn(HCOO)2(BPY)] | 4,4′-Bipyridine (BPY) | 300 W xenon lamp | 3 min | Stable in the dark for 10 months | 48 |
2.2 MOFs loaded with photoactive guests
Walton et al. reported the formation, structural and optical properties of a MOF constructed from Zn(II), terephthalate (pBDC) and triethylenediamine (TEDA).30 The MOF was loaded with a photochromic compound 1,2-bis(2,5-dimethyl-thien-3-yl)-perfluorocyclopentene (DTE, dithienylethene) using a thermal treatment method to give DMOF-1@DTE. When exposed to UV light radiation with a wavelength of 356 nm, the crystals exhibited a photochromic behavior which was evidenced by the change in the colour of the crystals from colourless to dark red. Intriguingly, the crystals were restored to their original colour upon radiation with visible light. Linear dichromism was observed in the irradiated crystals which confirmed the alignment of the DTE chromophores within the pores of the DMOF. Further studies confirmed that a single DMOF-1@DTE crystal could be repeatedly switched ‘on and off’ and that the photogeneration of the dark red species occurs more quickly than the subsequent back reaction to the colourless form.Guo and coworkers prepared an electron-transfer photochromic MOF compound with photoactive viologen cations as guests.38 The anionic framework [Cd3(BTC)2]n2n− was templated by (H2Bpy)2+ cations and lattice water molecules through weak interactions (BTC = 1,2,4,5-benzenetetracarboxylate, Bpy = 4,4′-bipyridine). The compound underwent a colour change from pale yellow to blue upon continuous irradiation with a 300 W xenon lamp at room temperature in air. After coloration, two new absorption bands around 393 and 602 nm appeared in the diffuse reflectance (DR) spectrum (Fig. 5) characteristic of N,N′-disubstituted-4,4′-bipyridinium monoradicals. ESR studies confirmed that the illumination resulted in the reduction of the (H2Bpy)2+ cations and the formation of (H2Bpy)+˙ radicals. The authors suggest that the oxygen atoms of the carboxylate moieties act as electron donors. A long lived charge-separated state was observed which can be explained by effective shielding of radicals by the host–guest structure and the presence of π–π stacking interactions around the (H2Bpy)+˙ radicals.
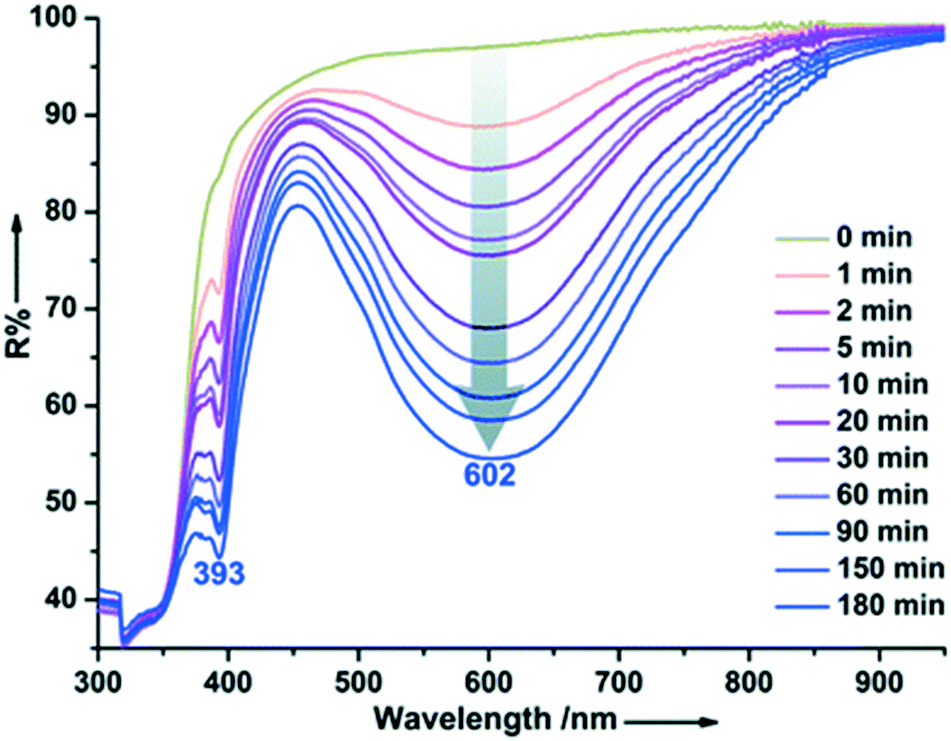 | ||
| Fig. 5 Time-dependent DR spectra of {(H2Bpy)[Cd3(BTC)2]·2H2O}n upon irradiation at room temperature in air. Reprinted from ref. 38 with permission from The Royal Society of Chemistry. | ||
A photoresponsive macromolecular framework [PV][Zn3(m-BDC)4]·H2O with a phenyl-substituted viologen dication as the guest templating-block and a zinc–carboxylate anionic framework as the host unit (PV = phenyl viologen, mBDC = 1,3-benzenedicarboxylate) was found to show photochromic behavior.39 Single crystal X-ray diffraction revealed that the phenyl viologen cation was located at the centre of a large channel interacting with the carboxyl groups distributed in the host framework. Irradiation of the MOF with light gives rise to a photoinduced electron transfer reaction that takes place from the donor carboxylate groups to the acceptor PV rings, accompanied by a quick colour change in the compound. Strong interactions between the electron rich host framework and the electron deficient guest cation paved way for intermolecular electron transfer.
The compound 2-(3′,3′-dimethyl-6-nitrospiro-[chromene-2,2′-indolin]-1'yl)acetic acid, (SP) was incorporated in a MOF via a two-step post-synthesis modification of the Zr-oxo nodes in MOF-808.40 MOF-808 was chosen for postsynthetic modification because of the high oxidation state of Zr4+ which enables strong metal–linker bonds, making MOF-808 highly chemically robust, and thus permitting the installation and further chemical modification of the spiropyran functional groups. Furthermore the pores in MOF-88 are able to accommodate the bulky SP guest molecules. The resulting host–guest inclusion compound showed photochromic behavior attributable to the isomerization of SP inside MOF-808.
2.3 MOFs prepared using naphthalenediide and photochromic switches
Lei and coworkers reported a photochromic Zn-based MOF derived from a naphthalenediide related tetrazole linker (NDI-ATZ) [Zn(NDI-ATZ)(DMF)2]n (NBU-3).41 Crystals of NBU-3 were reported to be sensitive to light and displayed a colour change from yellow to green (NBU-3b) when irradiated with sunlight for about 15 min. The green NBU-3b compound was very stable in air and could easily revert to the original colour when kept in a dark room for about 1 hour. PXRD studies confirmed that the structure of NBU-3 was identical to that of NBU-3b while the diffuse reflectance spectra exhibited different absorption bands (Fig. 6). These results confirmed that the photochromic behavior exhibited by the compound was not due to a structural transformation but rather to electronic transitions corresponding to n–π* and π–π* transitions of aromatic organic ligands.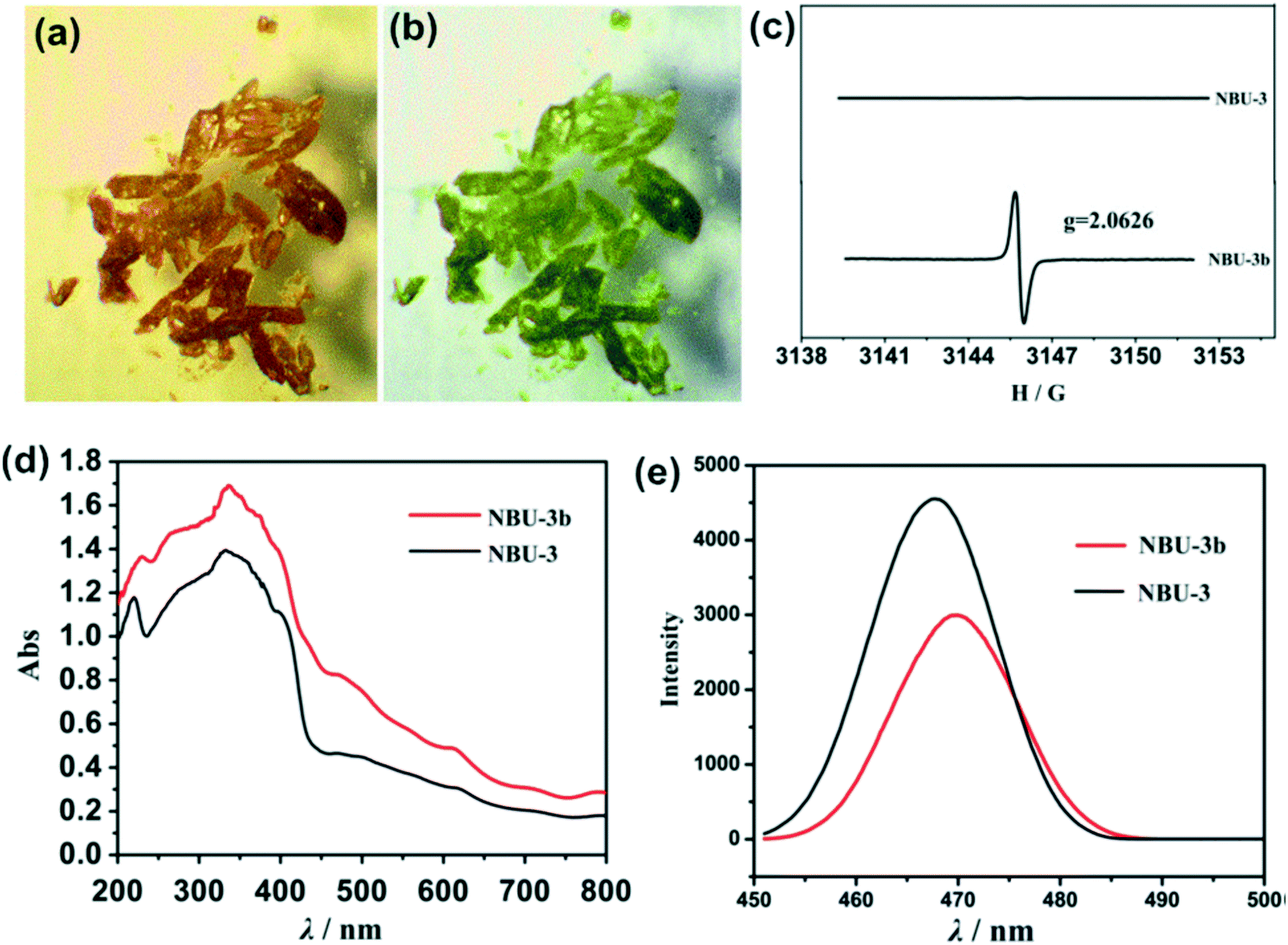 | ||
| Fig. 6 (a) and (b) Photochromic effect in NBU-3 single crystals (c) ESR spectra of NBU-3 and NBU-3b. (d) Diffuse-reflectance UV/vis spectra of NBU-3 and NBU-3b. (e) Emission spectra of NBU-3 and NBU-3b. Reprinted with permission from ref. 41. Copyright (2016) American Chemical Society. | ||
Park et al. reported a reversible photochromic porous coordination network (PC-PCN) assembled from Zn(II) and 1,2-bis(2-methyl-5-(pyridin-4-yl)thiophen-3-yl)cyclopent-1-ene (BPDTE) linker which served as a photochromic switch (Fig. 7).42 Colourless flake-shaped single crystals of the BPDTE-containing photochromic porous coordination network (PC-PCN), which had a light-yellow colour in the bulk sample, were obtained as illustrated in Fig. 7. When the open form of PC-PCN was exposed to UV light, blue crystals (purple in bulk) were clearly observed by the naked eye within 10 minutes. The photochromic effect is reversible upon irradiation of the crystals with visible light of 450 nm. This process occurred in a single crystal to single crystal manner as illustrated in Fig. 7b which demonstrated excellent structural reversibility of the BPDTE between the closed and open form.
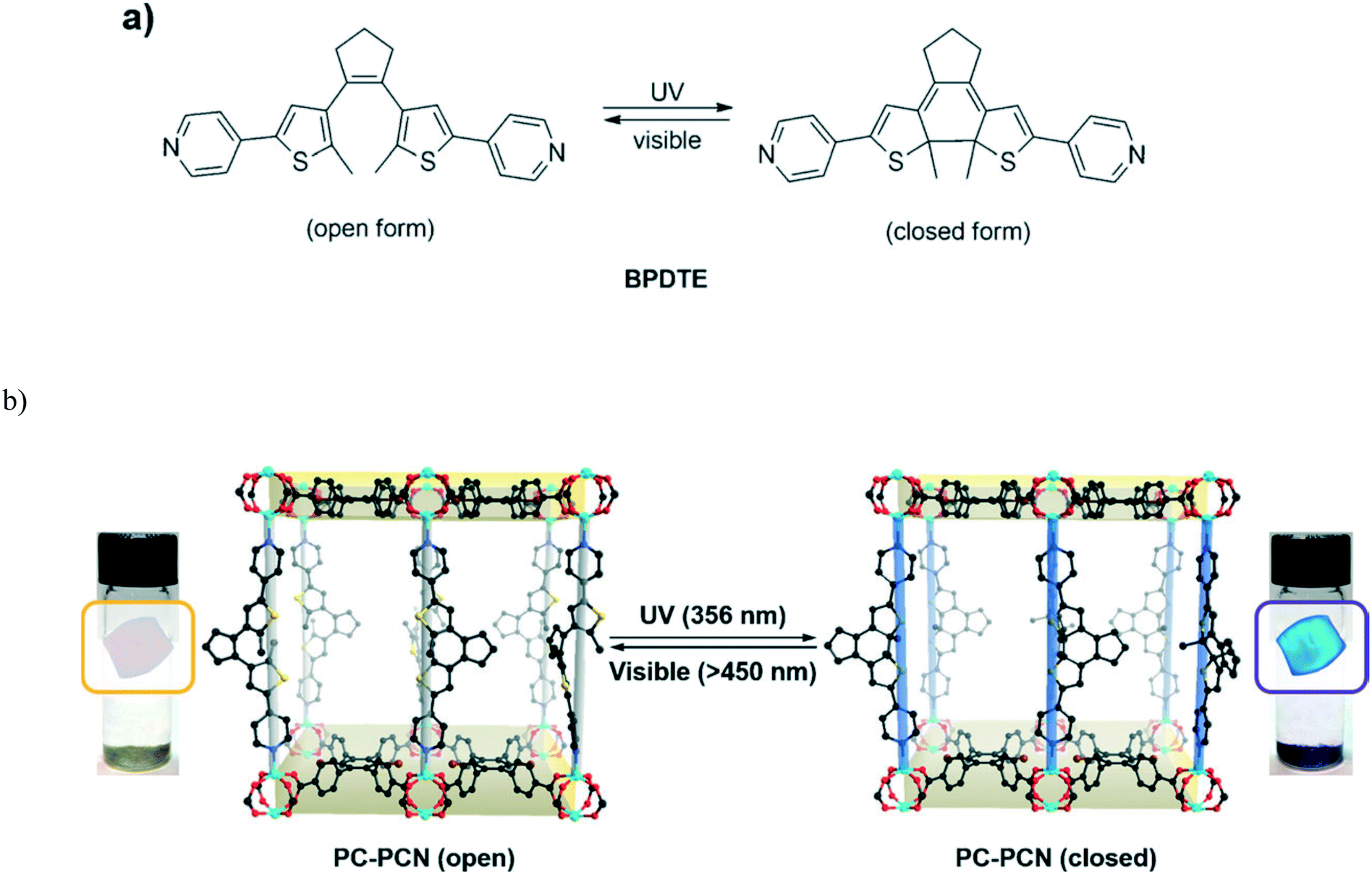 | ||
| Fig. 7 (a) Photoisomerization of BPDTE under UV and visible light. (b) Single crystal to single crystal photochromic behavior of the PC-PCN. Reproduced from ref. 42 with permission from John Wiley and Sons. | ||
A bis-carboxylate dithien-3-ylphenanthrene ligand was used to construct a photochromic responsive MOF.17 The photochromism of the linker was found to be reversible while that of the MOF could not return to the original colour upon irradiation visible light. It was concluded that the persistent colour after irradiation with visible light was attributed to the local environment of the ligand in the framework which suppressed the ring-opening reaction.
2.4 MOFs prepared using redox active compounds
Recently, a photo-active bipyridinium carboxylate linker 1-(3,5-dicarboxyphenyl)-4,4′-bipyridinium bromide (H2ipbpBr) was used to assemble two novel photochromic MOFs [Zn(ipbp)(H2O)]·NO3·H2O (1) and [Zn(ipbp)(H2O)2]·NO3·H2O (2).43 These two compounds were found to be photoactive to UV and visible light, and undergo visible colour change from light yellow to green. The dark green crystals of 1 were stable in air and displayed a reversible colour change to light yellow when they are kept in the dark for one day. The process was repeated for several times without a colour loss.UV-vis and EPR measurements were used to understand the origins of the photochromic process of 1 and 2. Fig. 8 shows two new absorption bands centred at 652 nm and 713 nm for 1 and 668 and 724 nm for 2 appear in the UV-vis spectrum after UV/visible light irradiation. The observed absorption bands were attributed to the generation of viologen radicals upon light irradiation. Further studies using in situ time-dependent EPR suggested that the photochromism of these compounds arises from the generation of the ipbp radicals. It is believed that upon irradiation of the crystals, electron transfer occurs from the O atoms of carboxylate groups to the N atoms of pyridinium units to generate ipbp radicals. Structural analysis revealed that the short distance between the adjacent carboxylate groups and the pyridinium ring of these compounds provide a suitable pathway for the electron transfer. Furthermore, the uncoordinated oxygen atoms of the carboxylate moiety ipbp ligands are also believed to favour the photoinduced electron transfer from carboxylate oxygen to pyridinium units. It was also noted that interpenetration of the framework facilitated the donor-accepter interaction. In a similar study, a zwitterionic bipyridinium carboxylate ligand 1-(4-carboxyphenyl)-4,4′-bipyridinium (hpc1) in the presence of 1,4-benzenedicarboxylate anions (BDC2−) and Zn2+ ions was used to construct two photochromic MOFs.44 EPR studies confirmed that the photochromism of the compounds originated from the generation of pyridinium radicals. Similar studies were performed by Zhang et al. and came to the same conclusions.45
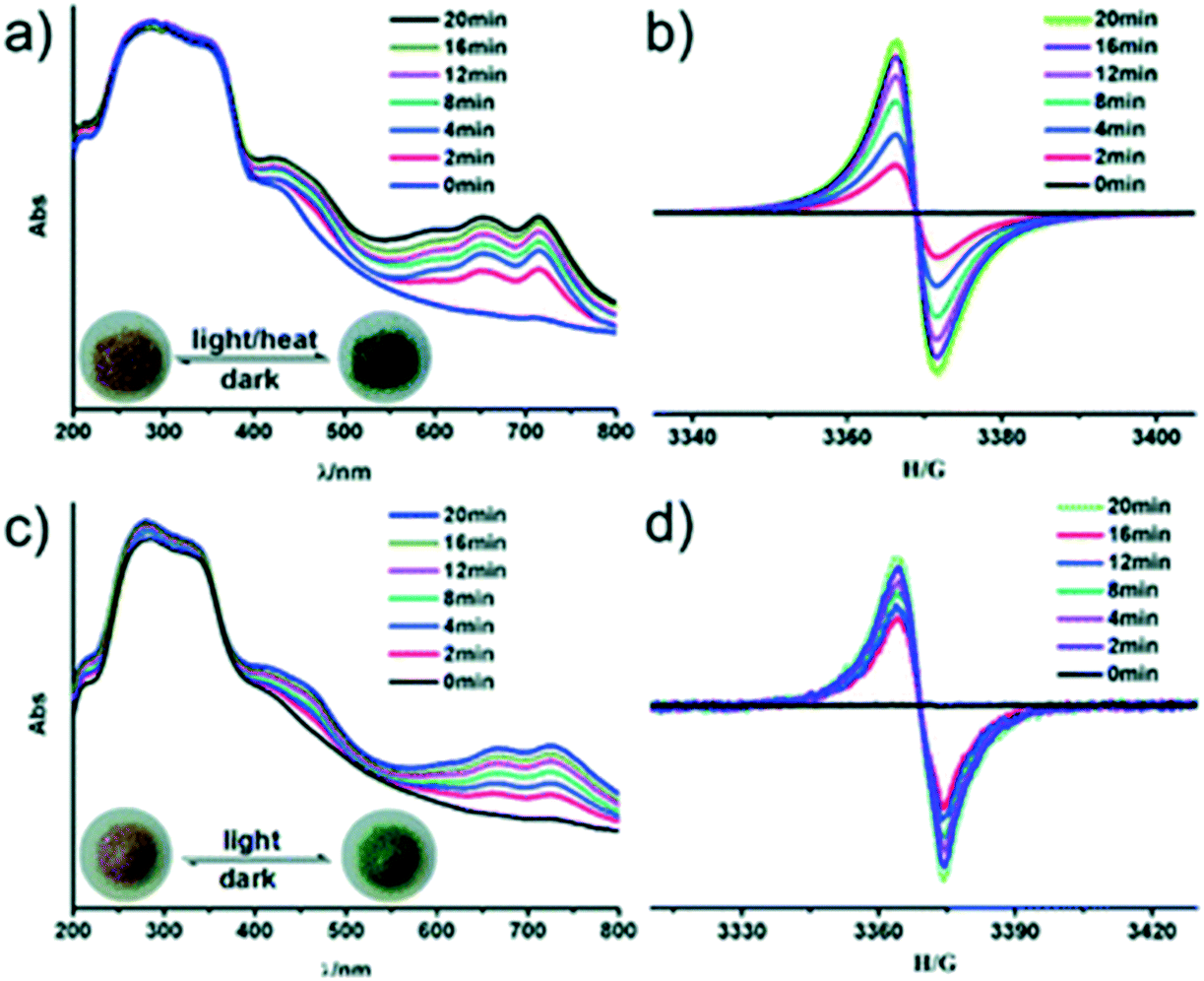 | ||
| Fig. 8 In situ time dependent UV/vis spectra and EPR signals of the original and UV irradiated samples for compounds 1 (a and b) and 2 (c and d), respectively. The insert photographs are crystals of before and after irradiation/heating. Reproduced from ref. 43 with permission from the Chinese Chemical Society (CCS), Peking University (PKU) and the Royal Society of Chemistry. | ||
A MOF constructed from the flexible zwitterionic viologen-tethered tetracarboxylate linker N,N′-bis(3,5-dicarboxylatobenzyl)-4,4′-bipyridinium (DCBPY2−): [Co3(DCBPY)(N3)4] showed reversible photochromism, which is related to the radical formation through photo-induced electron transfer from azide/carboxylate to viologen as confirmed by UV-vis, X-ray photoelectron, ESR and DFT calculations.19 It was observed that the crystal and molecular structure is maintained during the photochromic and the reverse processes. ESR and solid state UV-vis reflectance results suggested that the photochromism originated from the generation of organic free radicals via one-electron transfer, consistent with the redox activity. The colour of the compound was found to fade when exposed to air which was attributed to quenching of free radicals by oxygen. XPS and DFT calculations suggested that the electron transfer is most likely from azide and/or carboxylate to 4,4′-bipyridinium, provided that there are appropriate pathways. Structural analysis of the compound revealed that short contacts between the pyridinium and terminal azide nitrogen atoms could provide a favorable pathway for electron transfer, consistent with the XPS results and DFT calculations.
Cadmium, bipyridinium species N-(3-carboxyphenyl)-4,4′-bipyridinium (CPBPY) and terephthalate (pBDC) linkers were used to assemble a MOF with chemical composition [Cd(CPBPY)(pBDC)(H2O)]n.46 The compound possessed reversible photochromic behaviour in the solid state. An obvious colour change from pale-yellow to pale-blue was observed upon irradiation using a 365 nm UV light. Interestingly, the photo product was found to be stable for months even when irradiated by sunlight under oxygen atmosphere. The long lived charge separation state was ascribed to the close packing pattern of the compound as revealed by single crystal X-ray diffraction studies. Literature documents several studies on the viologen based linkers in MOFs and their associated photochromic effects.47,48
2.5 Applications of photochromic MOFs
Banerjee and co-workers demonstrated that photochromic MOFs can be used as photochromic inkless and erasable printing media. 1,4,5,8-Naphthalenediimide based MOFs were used to coat cellulose filter paper using a drop casting method.10 This was followed by surface smoothening with a glass slide as illustrated in Fig. 9. Drying was performed under vacuum to ensure that the coating adheres to the paper. The printing of the contents on the MOF coated paper was performed by controlling the incidence of sunlight through a stencil. The flexibility of the MOF coated paper was illustrated by mechanical deformation. Each of the characters was clearly distinguishable from its next neighbour. The colour contrast was so good to such an extent that the printed contents could be easily read. | ||
| Fig. 9 (a) Preparation of Mg–1,4,5,8-naphthalenediimide (Mg–NDI) coated paper; b) printing on the coated paper using stencil and sunlight, c) test for resolution of the printed content by the printing of letters on a 11.5 × 5.4 cm2 sheet; (d) mechanical deformation testing with Mg–NDI coated paper and (e) image showing ball and stick model of Mg–NDI structure on the Mg–NDI coated paper having a dimension of 14.9 × 8.1 cm2. Reprinted from ref. 10 with permission from the Royal Society of Chemistry. | ||
3. Solvatochromism/vapochromism
Solvatochromism/vapochromism may be defined as the ability of a chemical species to change colour upon inclusion of different solvents, as a result of the shift in the absorption spectrum of the material.24,49,50 This arises as a result of change in the energy gap between the ground state and the excited state, which is triggered by the interactions between the chromospheres and the solvent molecules. Solvatochromism is commonly reported in solution, but involves large quantities of solvent and is not of much utility in a sensing material. MOFs thus offer the potential of serving as solvent indicators in the solid state for small amounts of solvent. Organic linkers which can change conformation under the influence of solvent molecules may be of use in solvatochromic MOFs. For example, MOFs containing the bis(pyridyl) ligand shown in Fig. 10 can take up various solvents in their cavities; the hydrogen bonding interactions between solvent and linker give rise to a variety of colours in the solid material.51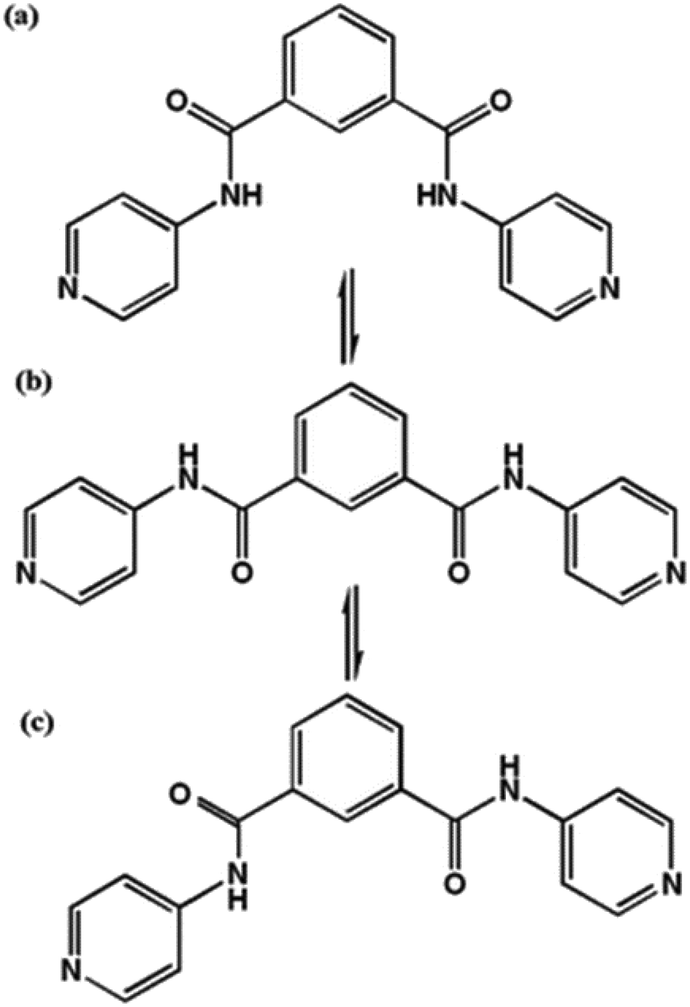 | ||
| Fig. 10 The cis- (a), intermediate- (b) and trans- (c) conformations of the bis(pyridyl) ligand, N1,N3-di(pyridin-4-yl)isophthalamide (DPPA), used to produce solvatochromic MOFs. | ||
Unlike conventional detection tools such as GC-MS or luminescence analysis that are either expensive or nonportable, solvatochromism provides a simpler and often easily visible method to detect organic molecules. Thus, the exploration of novel dual-functional MOFs with solvatochromic/vapochromic properties is of great important for further expanding the application of MOF sensors. Hereafter we will use the more generic term solvatochromic. Table 2 provides a summary of the properties of the solvatochromic MOFs discussed in this review.
| MOF | Linkers | Solvents | Response time | Stability of product | Ref. |
|---|---|---|---|---|---|
| [Cu(DPPA)2Cl2] | N 1,N3-Di(pyridine-4-yl)isophthalamide (DPPA) | Water | Immediate | Stable | 51 |
| [Ni(DPPA)2Cl2] | N 1,N3-Di(pyridine-4-yl)isophthalamide (DPPA) | Water | Immediate | Reversible | 51 |
| Mg–NDI | N,N′-Bis(5-isophthalic acid)naphthalenediimide (H4BINDI) | Polar organic solvents, amines, aromatic solvents | Rapid | Reversible | 57 |
| [Co(44pba)2] | 4-(4-Pyridyl)benzoate (44pba) | Water, polar organic solvents | Rapid | Reversible | 59 |
| [(WS4Cu4)I2(dptz)3] | 3,6-Di(pyridine-4-yl)-1,2,4,5-tetrazine (dptz) | Acetone, water, methanol, ethanol, acetonitrile, chloroform | Rapid | Reversible | 56 |
| [ZnNa2(μ2-H2O)2(H2O)2(TATAT)] | 5,5,5′-(1,3,5-Triazine-2,4,6-triyl)tris(azanediyl)triisophthalate (TATAT) | Acetone, ethanol | Several hours | Stable (not reversible) | 62 |
| [Cu2(4-pmpmd)2(CH3OH)4(opd)2] | N,N′-Bis(4-pyridylmethyl)phenyldiimide (4-pmpmd) and o-phthalic acid (opd) | Water, organic solvents | Immediate | Reversible | 63 |
| [Cu2(bzbp)2I2] | 1-Benzimidazolyl-3,5-bis(4-pyridyl)benzene (bzbp) | Polar aliphatic organic solvents | Response time dependent on solvent molecule size | Reversible | 66 |
| UiO-67-dmbpy | N,N′-Dimethyl-2.2′-bipyridinium (dmbpy) | Alkylamines | Rapid | Reversible | 67 |
3.1 Design aspects of solvatochromic MOFs
Solvatochromic behaviour in MOFs may arise from different mechanisms, including solvent polarity and specific solvent-chromosphere interactions, changes in the coordination geometry of the centre which leads to changes in the visible d–d energy transitions52–54 as well as metal-to-ligand-charge-transfer (MLCT) and π to π* transitions.55,56 Solvatochromic MOFs are relatively rare owing to the difficulty associated in achieving stable pores and solvatochromism in the materials.56 In designing MOFs with solvatochromic properties, the following approaches have been used: (1) use of solvatochromic ligands to link metal ions to form extended networks that are porous, (2) the use of flexible linkers that can easily form hydrogen bonds with solvent molecules (3) use of metal ions that are known to exhibit colour changes as a result of change in the coordination geometry, and (4) the use of carboxylate ligands that are capable of switching their binding mode. It is important to note that the framework must be porous to allow for the entry of the guest molecules. MOFs can be easily functionalised,22,57 therefore, it is possible to incorporate into the MOF appropriate recognition sites that can provide chemically specific receptor–analyte interactions. While strong interactions such as covalent bonds can be good for selectivity, they may be detrimental to stability and regeneration. To facilitate regeneration, the interactions should be reversible and must not destroy the frameworks. These interactions should lead to a visible colour change that can be observed by a naked eye. Fig. 11 displays some of the ligands used to construct the solvatochromic MOFs reviewed in this article.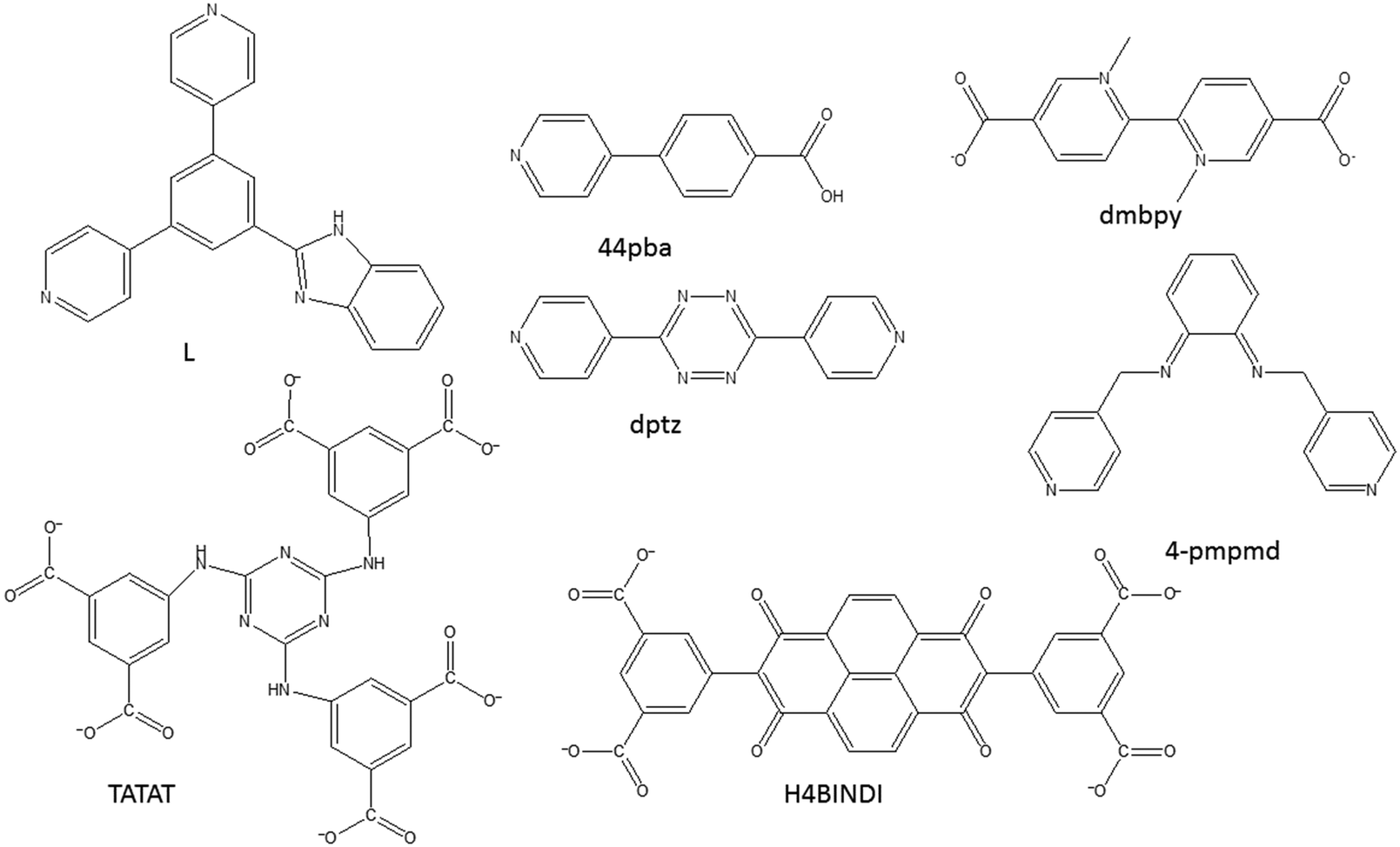 | ||
| Fig. 11 An illustration of some of the compounds used to construct solvatochromic MOFs discussed in this review. | ||
3.2 Solvatochromic MOFs
A magnesium based MOF Mg–NDI constructed using a naphtalenediimide linker (NDI) showed reversible solvatochromic behavior (Fig. 12).57 | ||
| Fig. 12 (a) ORTEP diagram of H4BINDI; (b) space filled diagram of Mg–NDI; (c) perspective view of a single pore and (d) perspective view of the extended structure of Mg–NDI. Reproduced from ref. 57 with permission from the Royal Society of Chemistry. | ||
Upon soaking the dark brown crystals of Mg–NDI in ethanol, the colour of the crystals changed to bright yellow. Further solvent exchange studies with DMSO, DEF, DMA, and MeCN generated different colours which were dependent on solvent polarity, as illustrated in Fig. 13. The colour change was observed to be reversible suggesting that the interaction between the solvent and the framework was short ranged and weak. UV-vis spectroscopic studies confirmed that the solvent containing Mg–NDI samples absorbed in the visible range and that the absorption maxima were consistent with the polarity of the solvents used. The Mg–NDI and the free H4BINDI ligand showed a strong absorption band at 370 nm attributable to the n–π* and π–π* transition of the aromatic carboxylate ligands. For the solvent containing Mg–NDI MOF, the UV-vis exhibited a gradual broadening of absorption band in the region 515–680 nm with respect to different solvent polarity. This may arise from the intermolecular electron-transfer transition from solvent captured in the MOF framework to BINDI linker. UV-vis data was used to calculate the band energy gaps for the solvents included in the MOF (Fig. 14). Except for ethanol and DMF, the band gap energies of other solvent incorporated MOFs were found to increase with the increment of the solvent polarity (1.87, 1.88, 1.91, and 1.92 eV) for DMA, DMSO, DEF, and MeCN, respectively.
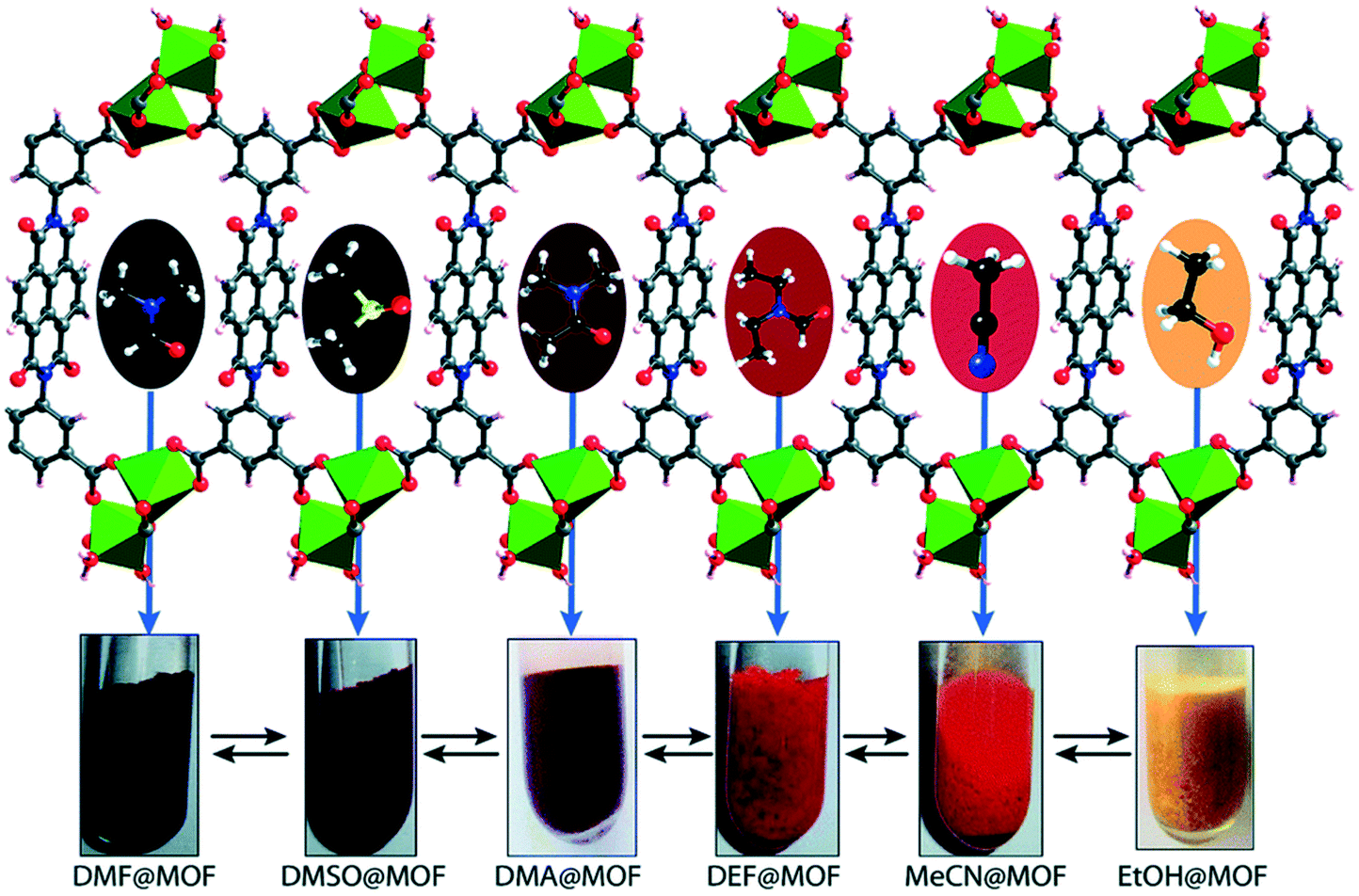 | ||
| Fig. 13 Schematic representation of Mg–NDI crystals showing different colours in different solvents. Reproduced from ref. 57 with permission from the Royal Society of Chemistry. | ||
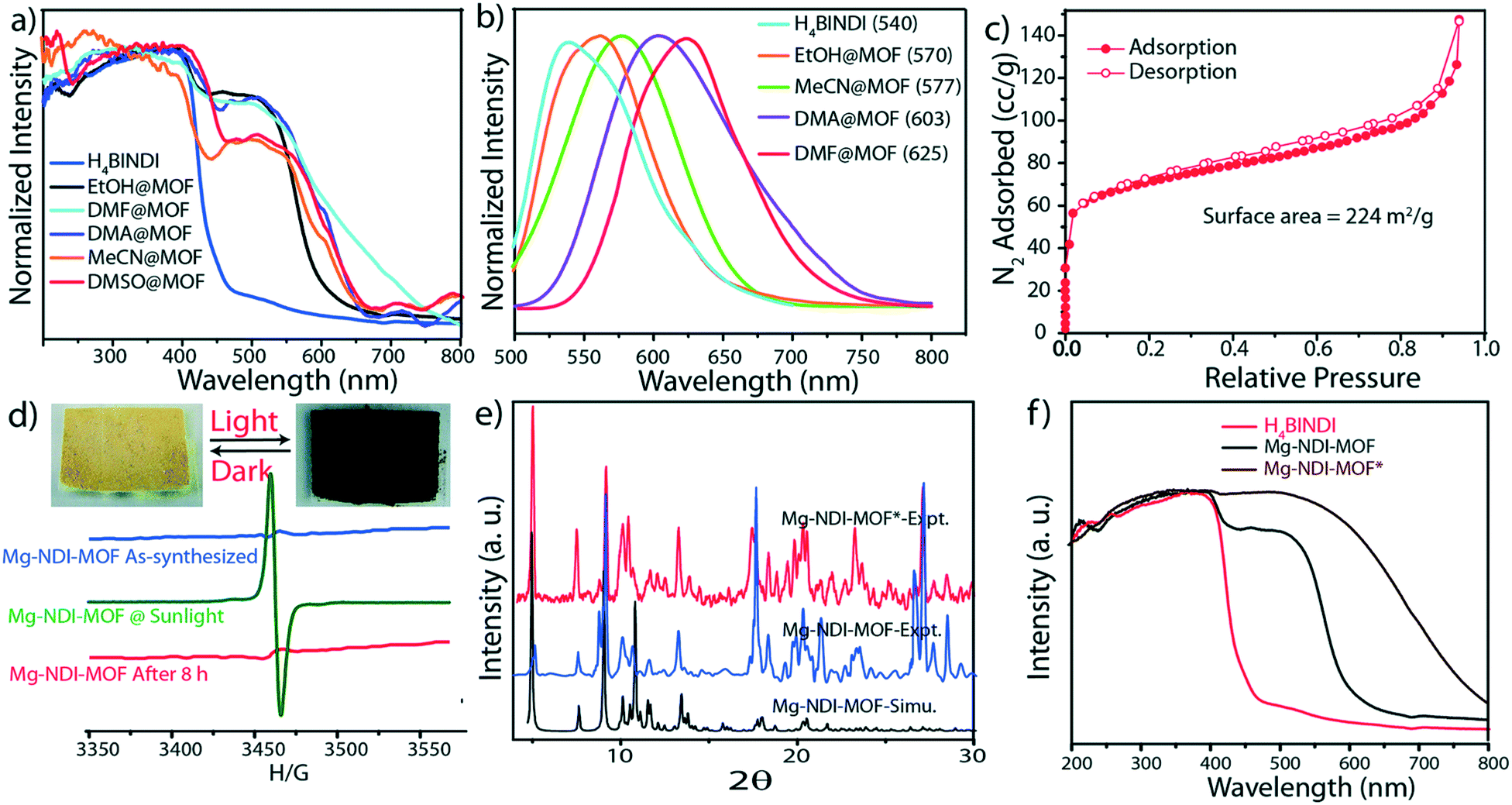 | ||
| Fig. 14 Solid state UV-vis spectra, (b) solid state photoluminescence of Mg–NDI crystals soaked in different solvents; (c) N2 adsorption isotherm for Mg–NDI; (d) ESR spectra of Mg–NDI crystals before and after irradiation of sunlight; (inset) photograph representing the reversible photochromic behavior of Mg–NDI MOF crystal in sunlight and dark; (e) PXRD patterns of Mg–NDI crystals before and after irradiation of sunlight; (f) solid state UV-vis spectra of H4BINDI (red), Mg–NDI (black) and Mg–NDI* (brown). Reproduced from ref. 57 with permission from the Royal Society of Chemistry. | ||
Further studies were performed to evaluate the ability of the Mg–NDI based MOF to detect organic amines. The presence of the chromophoric NDI moiety made Mg–NDI capable of sensing organic amines in the solid state.57 The MOF was found to selectively sense small sized organic amines by visual colour change. This can be explained by the fact that electron rich organic amines can form charge transfer complex with the NDI moieties within the framework giving rise to a change in colour. Treatment of Mg–NDI with various organic amines like aniline, hydrazine, ethylene diamine, triethylamine, dimethylamine, 1,3-propanediamine, ethylamine, methylamine gave rise to a distinct colour change (to black) over other functionalized analytes like chlorobenzene, toluene, benzene, phenol, 4-nitrophenol, nitrobenzene, 4-bromotoluene, etc. (Fig. 15). This colour change was extremely rapid and could be easily detected by the naked eye.
 | ||
| Fig. 15 (a) Photograph of selective detection of aniline over other aromatic functional molecules; (b) photograph showing the colour change of Mg–NDI samples in presence of different amines (0.1 M); (c) bar chart representation for quenching efficiency of Mg–NDI in presence of different amines (0.01 M) in EtOH; (d) reduction of PL emission intensities of Mg–NDI by gradual increase in concentration of aniline; (e) Stern–Volmer plot for Mg–NDI with different amines. Reproduced from ref. 57 with permission from the Royal Society of Chemistry. | ||
In our own work we designed several solvatochromic MOFs using pyridylbenzoate linkers with Co(II) salts.49,58–61 The reaction of 4-(4-pyridyl)benzoate (44pba) and Co(II) under solvothermal conditions yielded {[Co(44pba)2]·sol}n where sol represents the solvents that were included in the framework on the compound. Exposure of the compound to air resulted in a colour change of the crystals from purple to yellow. Soaking of the activated crystals of {[Co(44pba)2]·sol}n in other solvents such as ethanol, DMSO or methanol resulted in colour changes which could be attributed to hydrogen bonding interactions between the solvent molecules and the framework. Such interactions tend to change the crystal packing of the compound which results in an alteration of the d–d transitions. UV-vis studies of the included compounds were consistent with the observed colours. The network of {[Co(44pba)2]·sol}n could be reversibly transformed to other networks by soaking the compound in dry methanol for up to four weeks. These transformation occurred stepwise in a single crystal to single crystal manner and were easily followed by single crystal X-ray diffraction (Fig. 16). The dia network of {[Co(44pba)2]·sol} transformed to a qtz net via an intermediate pts net. These transformations were accompanied by colour changes as depicted in Fig. 16. Single crystal X-ray diffraction studies revealed that the changes in colour were attributed to the change in the coordination mode of the carboxylate group from the chelating to the modentate mode, as well as the effect of coordinating methanol molecules.
 | ||
| Fig. 16 An illustration of the transformations a 4-(4-pyridyl)benzoate-cobalt(II) MOF undergoes when soaked in dry methanol for an extended period.59 | ||
A similar study on the vapochromic behavior of the same compound20 found that the vapochromism observed on sorption of water was due to the change in geometry around the cobalt ion. The authors also suggested that a change in linker conformation may play a role in relieving the strain within the framework.
Zheng and coworkers prepared a nanotabular MOF {[(WS4Cu4)I2(dptz)3]·DMF}n (dptz = 3,6-di(pyridin-4-yl)-1,2,4,5-tetrazine, DMF = N,N-dimethylformamide), 1·DMF.56 Upon immersion of the dark red sample 1·DMF in CH3CN or CHCl3, the resulting inclusion compound 1·CH3CN changes to bright red colour, but 1·CHCl3 changes to black. The other solvents that were tested were acetone, water, methanol and ethanol. The resulting inclusion compounds changed to different colours depending on the solvent polarity. The band gaps of the inclusion compounds showed excellent linear correlation with the ENT values of solvents (Fig. 17, right). The best fits of band gaps against the ENT values were obtained by separately plotting nonhydroxylic solvents and hydroxylic solvents. The UV-vis spectra and photographs of these inclusion compounds are depicted in Fig. 17, the UV-vis absorption bands are well consistent with their colours. The solvatochromic effect of the dptz linker was evaluated by comparing the tests with an analogous MOF {[WS4Cu4(bpy)4][WS4Cu4(bpy)2I4]}n, (2, bpy = 4,4-bipyridine). The analogous MOF showed no solvatochromic effects upon guest exchange. The solvatochromic behaviour of {[(WS4Cu4)I2(dptz)3]·DMF}n and dptz linker was ascribed to MLCT and π to π* transitions respectively. The strong π-acceptor property and labile electronic structure to solvent polarity of the dptz linker in the nanotabular MOF played a crucial role in the solvatochromic behaviour exhibited by the nanotabular MOF. The authors concluded that the coupling of solvatochromism and porosity in a solid material has interesting prospects for the development of new sensing materials.
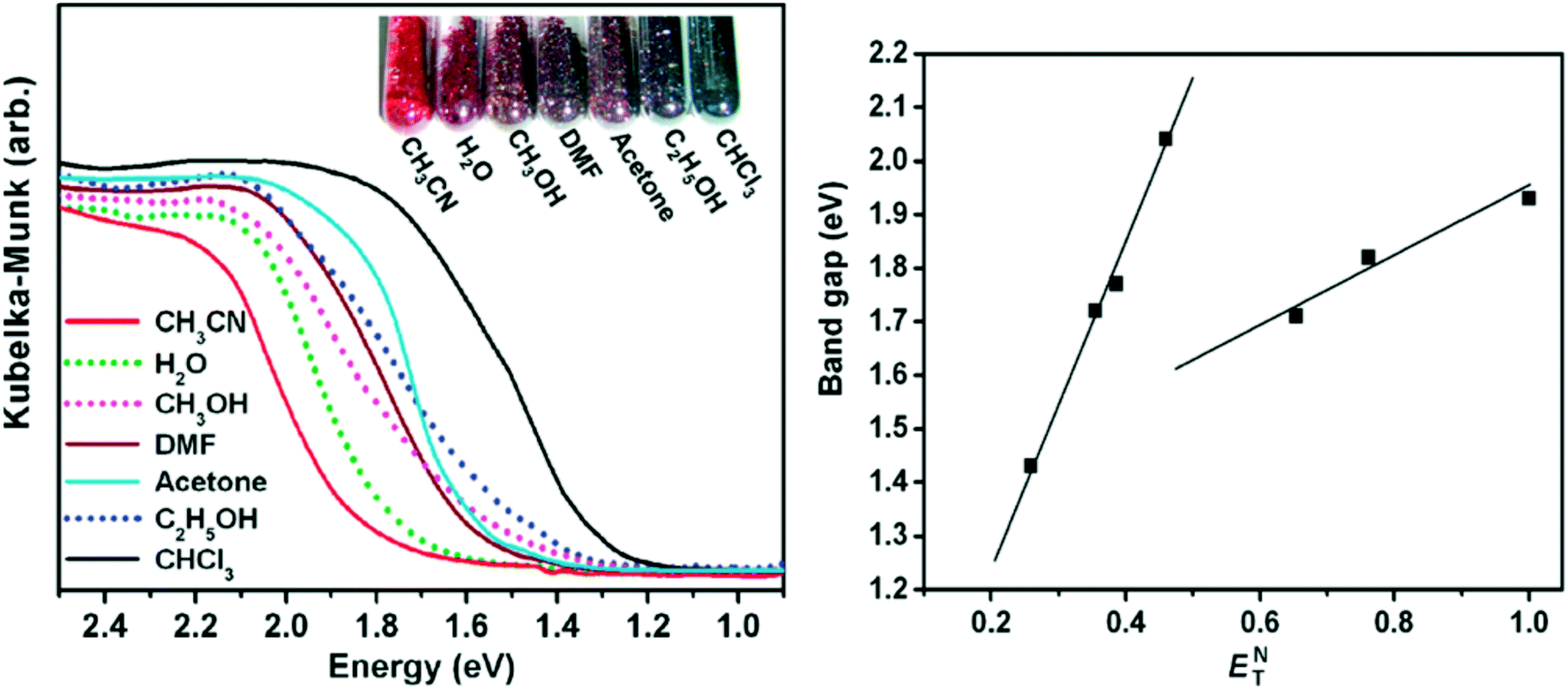 | ||
| Fig. 17 Left: The UV-vis spectra and photograph of the inclusion compounds 1⊃solvent. Right: Band gaps of the inclusion compounds against the solvent ET values. Reprinted with permission from ref. 56. Copyright (2011) American Chemical Society. | ||
Sun et al.62 evaluated the solvatochromic behaviour of a mesoporous chiral MOF constructed from a hexatopic ligand 5,5,5′-(1,3,5,-triazine-2,4,6-triyl)tris-(azanediyl)triisophthalate (TATAT) and zinc(II) ions. The solvothermal reaction product obtained was a 3D anionic MOF which has a channel volume of 69.8% of the unit cell volume. When the fresh samples of the 3D chiral network were immersed in acetone and ethanol, a visible colour change was observed from colourless to light yellow after 30 minutes and 5 hours and the intensity of the colour increased over time. Fig. 18 shows the photographs of the sample in the two different solvents acetone and ethanol.
 | ||
| Fig. 18 (a) Acetone loaded compound. (b) Ethanol loaded compound. (c) Infrared of solvent loaded compounds: black: as made, green: ethanol loaded, pink: acetone loaded. Reprinted from ref. 62 with permission from John Wiley and Sons. | ||
Removal of the coordinated solvent molecules and water molecules in the 2-fold interpenetrated MOF with composition {[Cu2(4-pmpmd)2(CH3OH)4(opd)2]·2H2O}n [4-pmpmd = N,N′-bis(4-pyridylmethyl)phenyldiimide; opd = o-phthalic acid] resulted in a colour change from blue to purple.63 Interestingly the blue colour could be regained on immersing of the activated phase to water or methanol molecules. It was noted that the other solvents such as ethanol, chloroform, acetone, benzene THF and DMSO failed to induce a colour change in the material which could be easily seen by the naked eye. When the UV-vis of the activated phase (purple in colour) was compared to the UV-vis of the complex formed after immersing it in water (blue in colour) there was about a 140 nm red shift of the absorption band which was induced by binding of the water molecules to the copper metal sites. The authors concluded that the activated phase of the synthesised material represents a simple copper(II)-based optical sensor for small hydroxylic molecules whose response is driven by a change of the metal-based d–d transitions on coordinating of the methanol or water to the metal centre.64,65
A porous CuI-MOF, [Cu2(bzpb)2I2] where bzpb = 1-benzimidazolyl-3,5-bis(4-pyridyl)benzene, can be used as a visual sensor for detecting small polar aliphatic volatile organic compounds (VOCs), such as alcohols, ketones, and halocarbons.66 After immersing the crystals of the as-made compound in methanol (MeOH), ethanol (EtOH), 2-propanol, n-propanol, ketones (acetone, 2-butanone, 2-pentanone and 3-pentanone), or chlorinated hydrocarbons (CH2Cl2 and CHCl3), colour changes could be detected by the naked eye (Fig. 19).
 | ||
| Fig. 19 Crystals of a CuI-MOF immersed in several solvents exhibiting visible colour change. Reprinted with permission from ref. 66. Copyright (2015) American Chemical Society. | ||
The rate of colour change depended on the size of the solvent molecules. This was explained based on the fact that smaller analytes could enter the pores more easily, which would lead to a faster colour response. Single crystal X-ray diffraction revealed that the guest molecules were confined in the pores of the framework without any covalent attachments to the metal ion. The colour change herein was attributed to an intermolecular electron-transfer transition between the host framework and encapsulated guests.
Single crystals are not always required or desirable in sensor materials. The ligand 2,2′-bipyridyl (bpy) in a Zr(IV) MOF (UiO-67-bpy) was N-methylated to generate N,N′-dimethyl-2,2′-bipyridinium (dmbpy).67 The N-methylated MOF (denoted as UiO-67-dmbpy) was deposited on a thin film and showed fast, selective and reversible change in colour in response to alkylamines. The authors concluded that the interaction between the generated 2,2′-bipyridinium moiety and the amine was responsible for the colour change. Solvatochromism of the UiO-67-dmbpy was ascribed to the electron-deficient bipyridinium moiety that forms a stable ground-state charge transfer (CT) complex with the electron-rich amine. The CT interaction not only provides an efficient driving force for recognition and absorption of amine molecules, but also leads to specific colour response.
4. Thermochromism
Thermochromic materials exhibit distinct colour changes at specific temperatures. These materials have technological applications in areas such as smart windows (used to improve the solar energy efficiency of houses and vehicles, reducing energy consumption and CO2 emissions), temperature sensors, colour filters and displays.68–70 The majority of studied thermochromic compounds are based on inorganic materials such as inorganic oxides, sulfides or halides but organic thermochromic compounds include compounds such as spirolactones, fluorans, and spiropyrans which are all known to change colour upon application of heat.71–74 In most of these materials, the thermochromic behaviour is correlated with a structural phase transition. A fundamental understanding of these materials is key to selection and design of new materials.4.1 Design aspects of Thermochromic MOFs
The possibility of a change in metal centre coordination geometry allows certain MOFs exhibit thermochromic properties.58,75,76 In designing MOFs with thermochromic behaviour, one might consider organic and inorganic components that are known to undergo some colour change with temperature change, such as certain cobalt and copper complexes. Organic linkers capable of changing their coordination modes, such as carboxylates, are good candidates for the design of thermochromic systems since the change in the coordination mode of chelating ligands affects the crystal field energy of the metal centre. In other situations loss of coordinated solvent molecules gives rise to visible colour changes.77 Studies have also confirmed that thermochromism in MOFs does not only arise from change in the ligand conformation. Disruption in the supramolecular environments such as hydrogen bonding interactions and π⋯π stacking as a result of lattice expansion triggers certain electronic transitions that are responsible for colour changes.75,76 Furthermore, changes in key dihedral angles are reported to trigger ligand to metal charge transfer electronic transitions. In other cases thermochromic effects have been attributed to spin cross over behaviour.78 There are few cases in which thermochromism has been attributed to the formation of stable radical species.18,19,79Table 3 provides some information about the thermochromic MOFs discussed below.| MOF | Linkers | Temperature of transition | Single crystal to single crystal? | Stability of product | Ref. |
|---|---|---|---|---|---|
| [Co(pBDC)(S-nia)] | Terephthalic acid (pBDC) and thionicotinamide (S-nia) | 105 °C, 4 h, vacuum | Yes | Regenerated on soaking in methanol | 80 |
| [Co(34pba)2] (two isomers) | 3-(4-Pyridyl)benzoate (34pba) | 281 °C and 347 °C | No | Irreversible | 58 |
| [Cu2(ETBC)(H2O)2] | 1,1′-Ethynebenzene-3,3′,5,5′-tetracarboxylate (EBTC) | 160 °C (as made) and 60 °C (guest-exchanged) | No | Not described | 81 |
| [Cu(pstp)2(SO3CF3)2] | 2-[5-(Pyridine-3-ylsulfanyl)-4H-1,2,4-triazol-3-yl]pyridine (pstp) | 50 °C and 80 °C | Yes | Reversible on cooling | 82 |
| [Co(μ2-H2O)(pBDC)(H2O)(DMF)] | 1,4-Benzenedicarboxylate (pBDC) | 105 °C | Yes | Not described | 54 |
4.2 Thermochromic MOFs
The reaction of 1,4-benzenedicarboxylic acid (pBDC) with thionicotinamide (S-nia), and Co(II) resulted in a Co(II)-based 2D coordination network {[Co2(μ2-OH2)(pBDC)2(S-nia)2(H2O)(dmf)]·2(dmf)·(H2O)}n that undergoes a single-crystal-to single-crystal (SC–SC) transition in the solid state upon desolvation.80 Heating the compound at 105 °C for 4 h in vacuum resulted in the desolvated product [Co(pBDC)(S-nia)]n. The SC–SC transition is accompanied by a colour change from pink to dark-blue as illustrated in Fig. 20. Single crystal X-ray diffraction data revealed that all coordinating solvent molecules were removed from the two cobalt centres present. This gave rise to change in the coordination geometry from octahedral to a square pyramidal shape. When the dry form was soaked in methanol for 24 h, the crystals reverted to their original colour (Fig. 20c). However the quality of the single crystals was not good enough for structural elucidation. Further studies showed that all the methanol molecules were restored during the soaking process. | ||
| Fig. 20 Photographs of single crystals of compound {[Co2(μ2-OH2)(pBDC)2(S-nia)2(H2O)(dmf)]·2(dmf)·(H2O)}n (a), as made (b) dried to [Co(pBDC)(S-nia)]n (c). Regenerated. Reprinted from ref. 80 with permission from the Royal Society of Chemistry. | ||
Time-dependent density functional theory (TDFT) was used to probe the nature of these colour changes. This study gives accurate spectral predictions of organic chromophores and coordination complexes. Electronic structure analysis shows that both the as made and dry binuclear complexes absorbed in the 420 to 500 nm range due to the presence of metal to ligand charge-transfer (MLCT) states. The dry phase had two additional new absorption peaks appear in the 650–800 nm range.
Mehlana et al. studied the thermochromic behaviour of a bcu net {[Co(34pba)2]·DMF}n assembled from 3-(4-pyridyl)benzoate linker and Co(II) ions under solvothermal conditions.58 Upon heating the compound, the crystals changed colour from pink to blue. The change in colour was attributed to change in the coordination geometry of the Co(II) metal centre from octahedral to a tetrahedral which has an effect of altering the d–d electronic transitions. IR studies were performed to probe the nature of the binding mode of the carboxylate moiety before and after the colour change. The results obtained suggested a change in the mode of binding from chelating to monodentate as evidenced by an increase in the magnitude of separation between the asymmetric and symmetric carboxylate stretches. In a related study, 3-(4-pyridyl)benzoate and Co(II) were used to construct a 3D hydrogen bonded discrete complex with composition {Co(34pba)2(H2O)4}n.75 The crystals of the discrete complex displayed three distinct colour changes (Fig. 21).
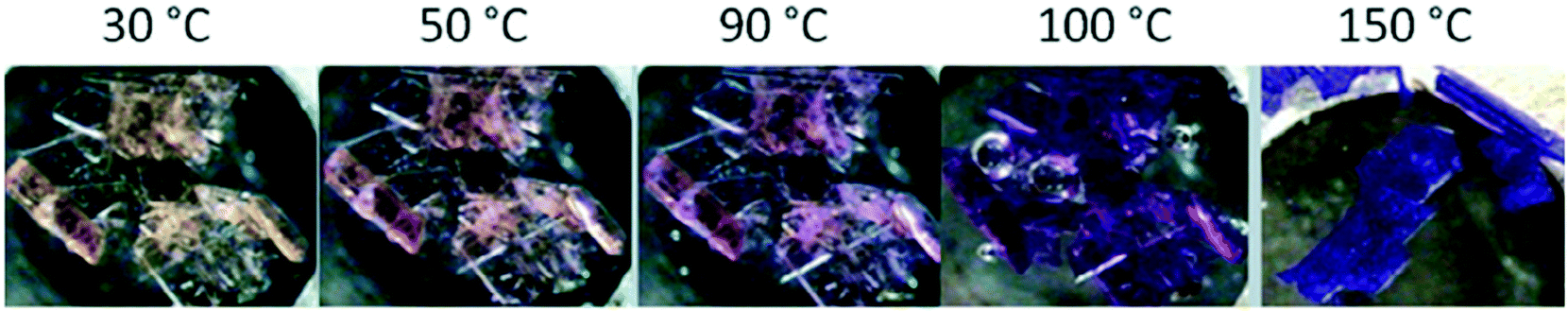 | ||
| Fig. 21 Crystals of {Co(34pba)2(H2O)4}n undergoing a colour change from yellow to dark pink and finally turning blue.75 | ||
To understand the origins of the observed colour changes, variable temperature PXRD studies were used to deduce the possible structural transformations occurring during the heating process. The results obtained showed a decrease in the lattice parameters along the a axis which may be attributed to changes in the Co–N bond distance or an alteration in the dihedral angles between the phenyl and the pyridyl rings. Furthermore, the decrease in the length of the b-axis has an effect of decreasing the distances of the π to π interactions. As opposed to the a- and b-axes which decrease with increase in temperature, the c-axis lengthened on heating suggesting that the hydrogen bonding interactions were weakened along this direction. It follows that thermochromic behaviour observed in the discrete complex within a temperature range of 30–120 °C may be attributed to the disruption of the hydrogen-bonding interaction and face-to-face π to π stacking caused by movement of the 1D chains upon application of heat. Disruptions of this nature may then trigger π to π transitions resulting in the observed colour changes (yellow to pink), although we may not rule the possibility of metal to ligand charge transfer. Above 150 °C (blue), thermochromism is ascribed to cooperative movement of atoms which involves loss of water molecules from the coordination sphere of the Co(II) ion followed by coordination of the carboxylate group to the metal centre. This has the effect of altering the crystal field around the Co(II) which gives rise to change in the visible d–d energy transitions.
A guest dependant thermochromic MOF was reported by Jin and co-workers.81 This MOF is composed of paddle-wheel-type dinuclear Cu2 secondary units and 1,1′-ethynebenzene-3,3′,5,5′-tetracarboxylate (EBTC4−) linkers formulated as Cu2(EBTC)(H2O)2[G] (G = guest molecules and represents DMF, DMSO and H2O). The guest molecules could be exchanged by soaking the crystals in methanol for 24 h to give [Cu2(EBTC)(H2O)2][(H2O)x(CH3OH)y]∞. Fig. 22 shows the crystals of the as made compound Cu2(EBTC)(H2O)2[G] (top) and the guest exchanged [Cu2(EBTC)(H2O)2][(H2O)x(CH3OH)y]∞ (bottom) exhibiting thermochromic behaviour.
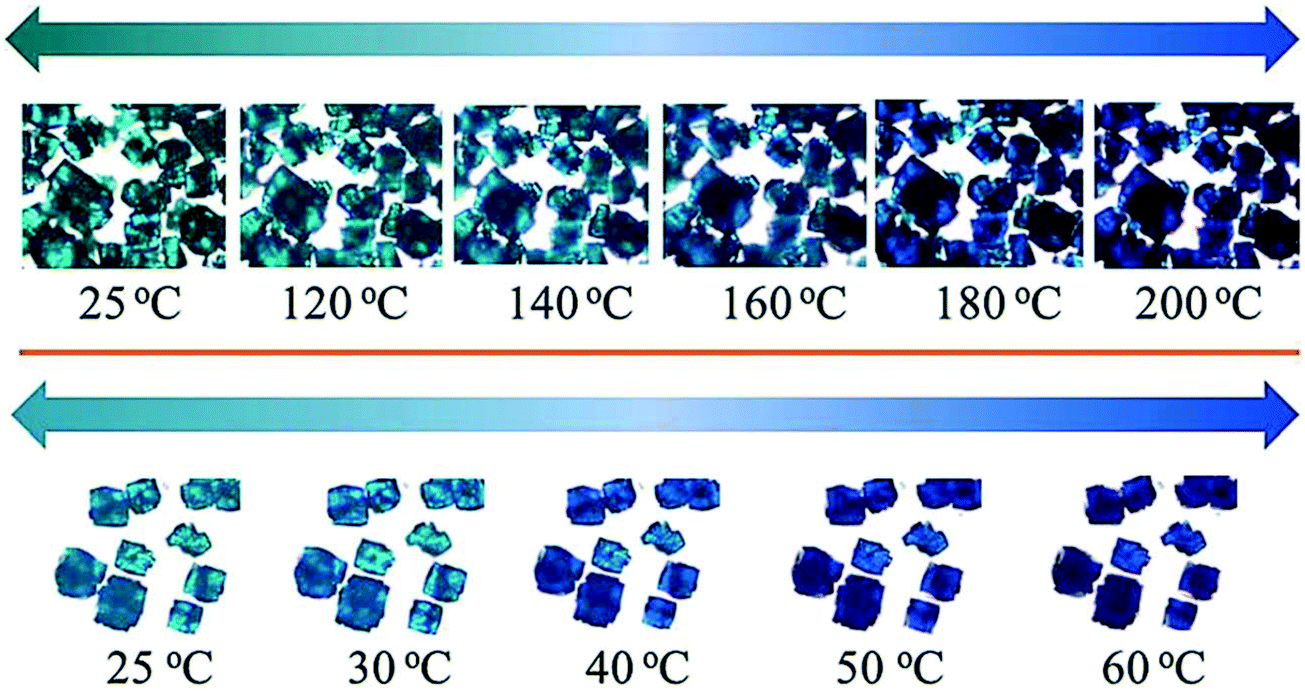 | ||
| Fig. 22 Images of crystals of the Cu2(EBTC)(H2O)2 MOFs at selected temperatures. Top: As made, bottom: guest exchanged. Reprinted from ref. 81 with permission from the Royal Society of Chemistry. | ||
Crystals of the as-made compound were transformed from greenish-turquoise (25 °C) to turquoise (until 140 °C) and light blue (160 °C) upon heating. In contrast, the guest-exchanged crystals' thermochromic effect was observed in a narrow temperature range. The colour changed from green-turquoise (25 °C), light blue, and finally navy blue (60 °C) upon raising temperature. It was noted that the colour change was fully reversible for both the as made and guest exchanged crystals. To understand the origins of these colour changes, variable temperature PXRD patterns of the as-made and guest-exchanged crystals was inspected at various temperatures. The PXRD for the guest-exchanged crystals did not show any significant change due to poor signal to noise ratio. However the as-made compound showed a shift to lower 2θ angles with increasing temperature for the (2 0 2) and (3 0 3) reflections (Fig. 23). This shift, particularly noticeable above 150 °C, indicates an expansion in the crystal lattice with temperature rise. It follows that the thermochromic effect originates from the lengthening of the Cu–O bond with increasing temperature which gives rise to a change of the crystal field around the Cu(II) ions.
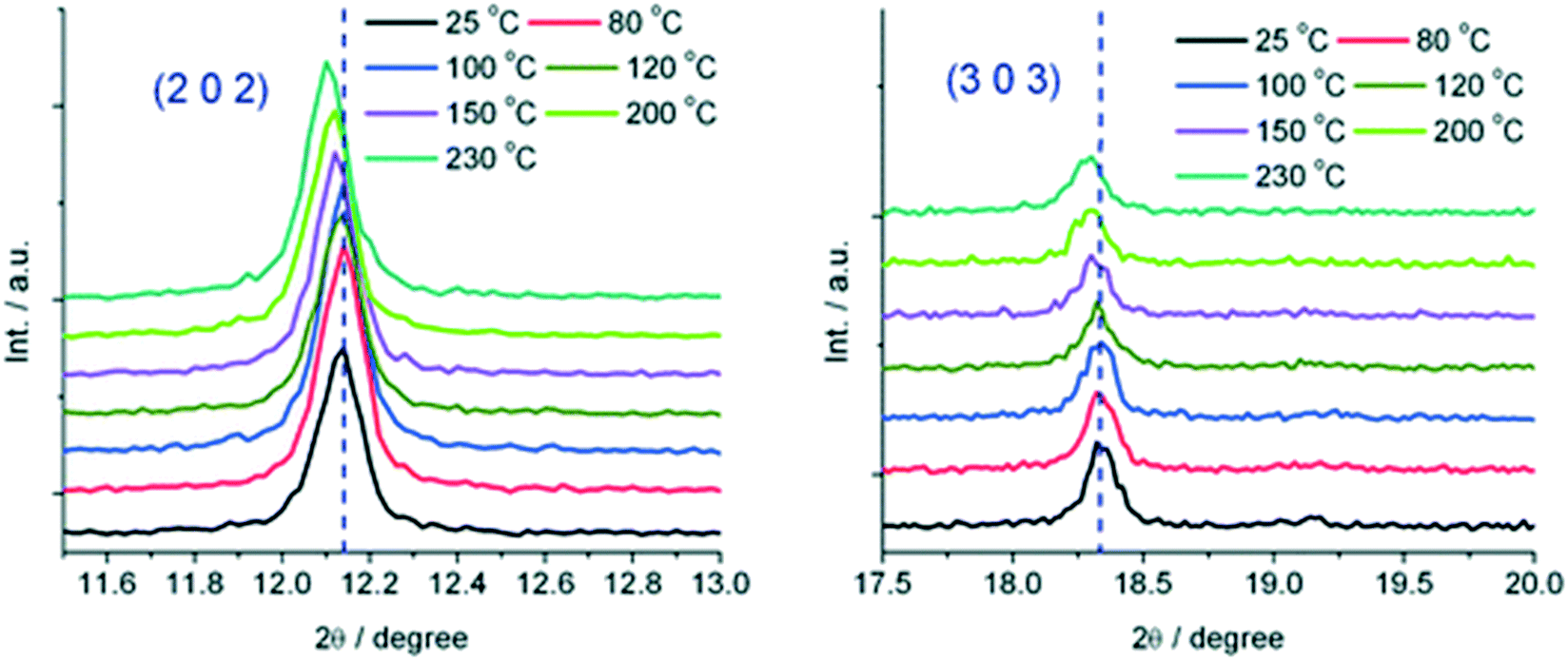 | ||
| Fig. 23 Temperature dependent reflections (2 0 2) and (3 0 3) at selected temperatures. Reprinted from ref. 81 with permission from the Royal Society of Chemistry. | ||
The reaction of a triazole-bridged asymmetric ligand, 2-[5-(pyridin-3-ylsulfanyl)-4H-1,2,4-triazol-3-yl]pyridine (pstp), with Cu(SO3CF3) produced air-stable sky-blue crystals with chemical composition (Cu(pstp)2(SO3CF3)2) (Fig. 24).82 The crystals transformed from blue to blue green at 50 °C and then to green at 80 °C. The colour change was fully reversible upon cooling. No guest molecules were found in the as-made crystals which suggested that the thermochromic effect was not guest driven. Single crystal X-ray diffraction studies were used to probe the structural changes that occurred during the colour transformation. Periodic changes in the Cu–N bonds were observed. These changes were responsible changes in the dihedral angle between the benzyl and triazole ring. The authors concluded that the changes in the dihedral angle facilitates the LMCT (ligand to metal charge transfer) process, which was supported by their UV-vis spectra (Fig. 24). DFT calculations also confirmed that electronic transition mainly occurs on the ligand and Cu(II) ion and the transferred electron is located mainly on the ligand.
 | ||
| Fig. 24 Top: Crystal pictures of Cu(pstp)2(SO3CF3)21, and the corresponding thermochromic compounds 2, 3 and 4. Bottom: Solid-state temperature dependence of UV-vis spectra of 1 (blue line), 2 (blue-green line), 3 (green line) and 4 (dashed line). Reprinted from ref. 82 with permission from the Royal Society of Chemistry. | ||
Fonari and coworkers reported a three-dimensional MOF, {[Co(μ2-H2O)(pBDC)(H2O)(DMF)]·0.5H2O}n which showed a colour change from light brown to pink upon evacuation of guest molecules at 105 °C.54 The process happened in single crystal to single crystal fashion and was accompanied by a change in the crystal system from triclinic centrosymmetric space group to a orthorhombic Imma. Similar studies were performed by the same group using a cobalt MOF assembled from dicarboxylate and nicotinamide-like linkers, where desorption of guest DMF at 105 °C led to similar observations.83
5. Piezochromic effects
Piezochromism is the change in colour of a solid under compression or change in pressure.84,85 Traditionally, this phenomenon can result from three mechanisms (1) the colour of a solid results from the absorption of light in selected regions of the visible spectrum by excitation of an electron from the ground electronic state to a higher level. If the two electronic energy levels are perturbed differently by pressure, compression results in a colour change; (2) a discontinuous change of colour occurs when a crystalline solid undergoes a first-order phase transition from one crystal structure to another; (3) change in the molecular geometry of the molecules that make up the solid. Piezochromic behavior which is based on modification changes of inorganic crystalline materials, such as LiF or NaCl monocrystals have been studied for several decades. It is well know that the transition from the green α- into the red γ-modification of CuMoO4 requires a pressure of 2.5 kbar. Palladium complexes have also been widely studied and are reported to require pressures ranging from 14 to 65 kbar. Piezochromic effects in organic polymer materials have also been described.86 However, the high pressure which is necessary for the modification changes makes these materials unsuitable as pressure sensors in ordinary life.5.1 Design principles for Piezochromic MOFs
Piezochromic MOFs offer new qualities, owing to their relative softness and to the presence of open pores enabling the adsorption of pressure-transmitting media and their direct interactions with chromophores in the bulk of the crystal. Porosity and flexibility are important properties that can be exploited to design piezochromic MOFs. Unlike traditional hard materials which require high pressure for a colour change to occur, piezochromism is predicted to occur at low relatively low pressures in MOFs. Despite the advantages offered by these materials, there has been only one reported case of a piezochromic MOF, and another of piezochromism in a 1D coordination polymer.87,88 Unlike traditional organic and inorganic compounds, piezochromism in MOFs could arise from several mechanisms in addition to those discussed above. For example guest migration induced by pressure changes could lead to visible colour changes due to increase in interaction between the host and guest. Compression does not only lead to distortion of the coordination environment of the metal centre but may also give rise to change in the ligand conformation and trigger electronic transitions.5.2 Piezochromic MOFs
A porous MOF, [Co(pBDC)(TEDA)(H2O)], denoted as AMU-1 was built from Co(II) linked by 1,4-benzenedicarboxylate (pBDC) anions and 1,4-diazabicyclo[2.2.2]octane (TEDA) molecules and displayed visible colour changes when compressed.87 This phenomenon was confirmed by UV-vis spectroscopy which displayed changes the absorption spectra with increase in pressure. Single crystal X-ray diffraction was used to understand the origins of piezochromism in AMU-1. The observed piezochromism of AMU-1 was ascribed to the low symmetry of the crystals and with the distortions of pseudo-octahedral coordination of cations Co2+, located in the highly asymmetric crystal field composed of four types of ligands. It was noted that high pressure increased the interactions among the pBDC, TEDA and water linkers embedded in the low-symmetry crystal environment which in turn enhanced the distortions of the pseudo-octahedral crystal field around Co2+. These distortions gave rise to the reduction in the crystal symmetry from monoclinic to triclinic (Fig. 25).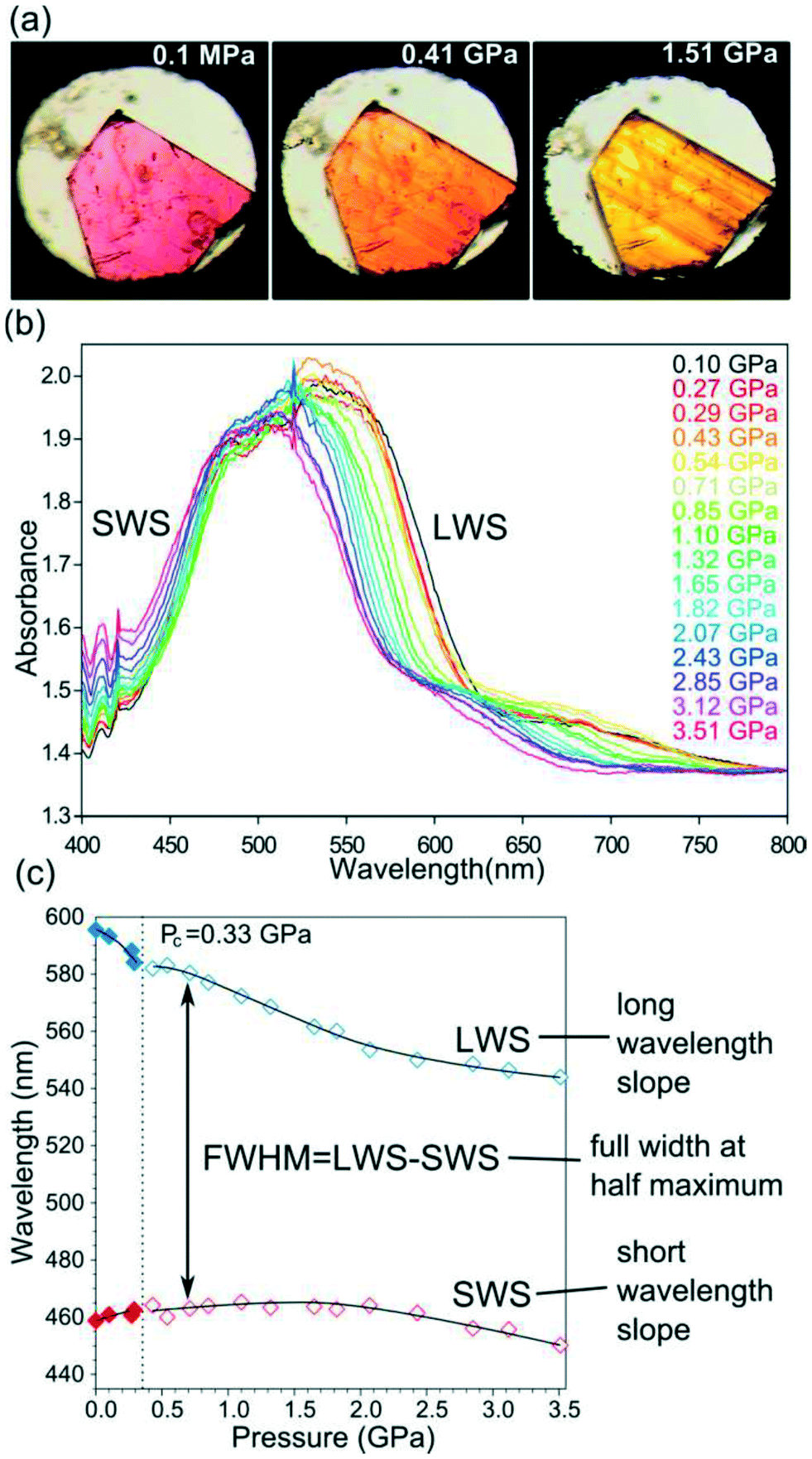 | ||
| Fig. 25 An illustration of piezochromism in AMU-1: (a) a single crystal showing a colour transformation from purple (phase α) to orange and yellow (phase β); (b) selected high-pressure VIS spectra of AMU-1; and (c) the plot shows shifts of LWS and SWS of the dominant peak fitted with polynomial functions (cf. SI). Reprinted with permission from ref. 87 Copyright (2017) American Chemical Society. | ||
A similar study was conducted using 1D coordination polymer [CoCl2(bpp)] (bpp = 1,3-bis(4-pyridyl)propane) which showed piezochromic reversible transformations at 1.93 GPa from blue phase α to green phase β, and at 2.39 GPa to colourless phase γ.88 The change from the α phase to the β phase is accompanied by considerable shortening of Co⋯Cl contacts between neighbouring chains. As pressure increased to 2.39 GPa the γ phase was formed and single crystal X-ray diffraction revealed a change in the topology of the network from 1D to 2D. This was also accompanied by a change in the coordination geometry from tetrahedral to octahedral (Fig. 26).
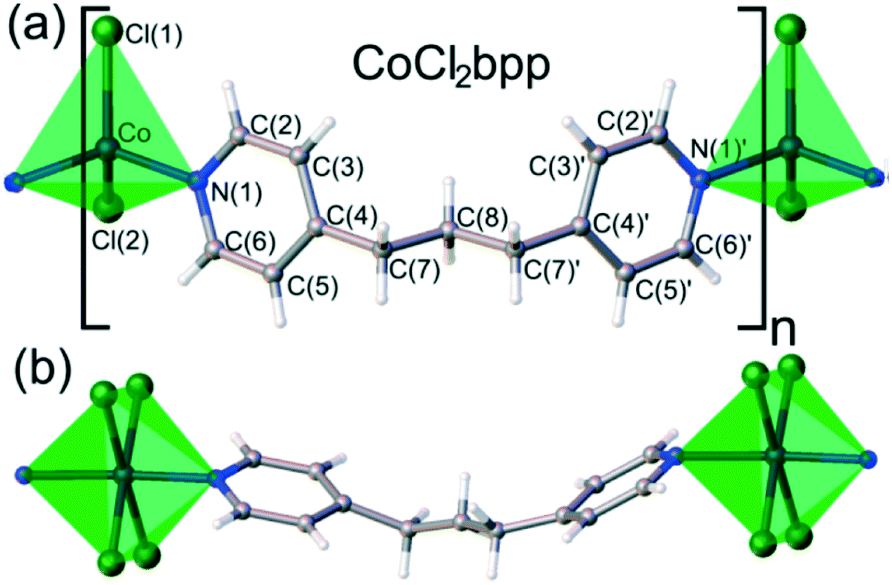 | ||
| Fig. 26 a) The α and β phase of [CoCl2(bpp)] which have a tetrahedral coordination geometry, the Co2+ cation is coordinated by two chloride anions and two pyridine rings b) the γ phase which has an octahedrally coordinated to by four chloride anions and two pyridine rings. Reprinted with permission from ref. 88 Copyright (2017) American Chemical Society. | ||
The authors concluded that that the piezochromism of [CoCl2(bpp)] is chiefly associated with the phase transitions of the crystal and with the abrupt changes in the Co-cation coordination, whereas only very gentle monotonic changes take place within the phases.
6. Conclusions and perspectives
In this review we have discussed four types of chromic MOFs, namely photochromic, solvatochromic, thermochromic and piezochromic. Single crystal diffraction remains the most powerful tool for studying all of these systems, as it allows the elucidation of supramolecular interactions playing a role in the chromism. In addition, a variety of complementary techniques play an important role in deepening the understanding of the phase and electronic transitions occurring. EPR and UV-visible spectroscopy have been widely used, along with IR spectroscopy and powder X-ray diffraction, which is particularly useful for following phase changes occurring in materials unsuited to single crystal diffraction studies. The increasing use of computational methods is anticipated to provide insights otherwise not readily accessible.Through careful selection of the starting materials and a better understanding of crystal engineering, chromic MOFs can be designed and fine-tuned to achieve desirable properties. In some cases, chromic MOFs have been discovered by design rather than by chance thanks to developments in the areas of soft chemistry, nanochemistry, and crystal engineering. Some challenges remain, for example solvatochromic MOFs are limited by the difficulty in achieving solvatochromic and stable pores in a single MOF. In addition, the majority of the reported solvatochromic MOFs are easily poisoned by water which further limits their practical applications such as detecting volatile organic compounds.20,89,90 Known photochromic MOFs do not perform well enough to be utilized as practical devices because of their poor conversion ratios and duration as well as their low photoresponse, sensitivity, and nondestructive readout capability. Recent studies have demonstrated that photoresponse in host–guest systems can be improved by increasing the interactions between the electron rich framework and electron deficient guest molecules. The introduction of phenyl substituted viologen cations into the pores of a MOF formed by metal–carboxylate connections appears favourable for building a stable π-conjugation electron acceptor unit. This design strategy is of great advantage as it increases the electron withdrawing properties of the viologen cation and the stability of the resultant viologen radical through the π-conjugation molecular system, which tunes the photoactive properties of the resulting supramolecular assembly. In other systems interpenetration has been used to increase the photoresponse of the MOFs as it provides a suitable path way for electron transfer by reducing the distance between the donor and acceptor atoms. On the other hand design systems based on incorporating photochromic ligands which undergo cis–trans isomerisation face some challenges. For example, interpenetration my hinder the change in the conformation of the linker rendering the photochromic process non-reversible. Furthermore, there must be enough space within the pores of the framework to allow for conformational change of the photochromic linker. All these aspects must be taken into consideration for design purposes.
Thermochromism in MOFs has been attributed principally to the change in the geometry of the metal centre but recent studies have shown that thermochromism can also arise due to disruption of the supramolecular environment caused by changes in π–π and hydrogen bonding interactions. There are a few reports which attribute thermochromism to the generation of radicals upon heating.
Piezochromism has been well studied in organic and inorganic systems. However, such systems require high pressure for the phenomenon to be observed. This limits the practical applications of these materials as pressure sensors. Flexibility coupled with porosity may allow MOFs to exhibit piezochromic effects at low pressure, making them suitable to be used as pressure sensors. While reported examples are rare, it should be possible to predict and design new piezochromic MOFs, giving rise to another burgeoning area of study in the field of chromic MOFs and related materials.
Acknowledgements
Funding from the South African National Research Foundation (grant number 90495) is gratefully acknowledged.References
- G. Mehlana, G. Ramon and S. A. Bourne, Z. Kristallogr. - Cryst. Mater., 2013, 228, 318–322 CrossRef CAS
.
- J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef CAS
- P. Falcaro, R. Ricco, C. M. Doherty, K. Liang, A. J. Hill and M. J. Styles, Chem. Soc. Rev., 2014, 43, 5513–5560 RSC
- S. Furukawa, J. Reboul, S. Diring, K. Sumida and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5700–5734 RSC
- S. Ma and H.-C. Zhou, Chem. Commun., 2010, 46, 44–53 RSC
- Z. Xie, L. Ma, K. E. DeKrafft, A. Jin and W. Lin, J. Am. Chem. Soc., 2010, 132, 922–923 CrossRef CAS PubMed
- G. Mehlana, S. A. Bourne and G. Ramon, CrystEngComm, 2014, 16, 8160 RSC
- X. Meng, B. Gui, D. Yuan, M. Zeller and C. Wang, Sci. Adv., 2016, 2, e1600480 Search PubMed
- S. Ou and C.-D. Wu, Inorg. Chem. Front., 2014, 1, 721–734 RSC
- B. Garai, A. Mallick and R. Banerjee, Chem. Sci., 2016, 7, 2195–2200 RSC
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed
- SciFinder Scholar, version 2017, Chemical Abstracts Service, Columbus, OH, 2017, (accessed 22 March 2017) Search PubMed.
- R. Huang, M. R. Hill, R. Babarao and N. V. Medhekar, J. Phys. Chem. C, 2016, 120, 16658–16667 CAS
- O. S. Bushuyev, T. Friščić and C. J. Barrett, CrystEngComm, 2016, 18, 7204–7211 RSC
- N. Yanai, T. Uemura, M. Inoue, R. Matsuda, T. Fukushima, M. Tsujimoto, S. Isoda and S. Kitagawa, J. Am. Chem. Soc., 2012, 134, 4501–4504 CrossRef CAS PubMed
- S. Castellanos, A. Goulet-hanssens, F. Zhao, A. Dikhtiarenko, A. Pustovarenko, S. Hecht, J. Gascon, F. Kapteijn and D. Bløger, Chem. – Eur. J., 2016, 22, 746–752 CrossRef CAS PubMed
- D. G. Patel, I. M. Walton, J. M. Cox, C. J. Gleason, D. R. Butzer and J. B. Benedict, Chem. Commun., 2014, 50, 2653–2656 RSC
- C. Tao, J. Wu, Y. Yan, C. Shi and J. Li, Inorg. Chem. Front., 2016, 3, 541–546 RSC
- T. Gong, X. Yang, J.-J. Fang, Q. Sui, F. G. Xi and E.-Q. Gao, ACS Appl. Mater. Interfaces, 2017, 9, 5503–5512 CAS
- M.-H. Zeng, Y. Tan, Y. He, Z. Yin, Q. Chen and M. Kurmoo, Inorg. Chem., 2013, 52, 2353–2360 CrossRef CAS PubMed
- J. D. Evans, C. J. Sumby and C. J. Doonan, Chem. Soc. Rev., 2014, 43, 5933–5951 RSC
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC
- F.-Y. Yi, D. Chen, M.-K. Wu, L. Han and H.-L. Jiang, ChemPlusChem, 2016, 81, 675–690 CrossRef CAS
- M.-S. Wang, G. Xu, Z.-J. Zhang and G.-C. Guo, Chem. Commun., 2010, 46, 361–376 RSC
- R. Pardo, M. Zayat and D. Levy, Chem. Soc. Rev., 2011, 40, 672–687 RSC
- D. H. Qu, Q. C. Wang, Q. W. Zhang, X. Ma and H. Tian, Chem. Rev., 2015, 115, 7543–7588 CrossRef CAS PubMed
- S. Helmy, F. A. Leibfarth, S. Oh, J. E. Poelma, C. J. Hawker, J. R. de Alaniz and J. Read, J. Am. Chem. Soc., 2014, 136, 8169–8172 CrossRef CAS PubMed
- G. Berkovic and V. Weiss, Chem. Rev., 2000, 100, 1741–1753 CrossRef CAS PubMed
- I. M. Walton, J. M. Cox, J. A. Coppin, C. M. Linderman, D. G. Patel and J. B. Benedict, Chem. Commun., 2013, 49, 8012–8014 RSC
- H.-Y. Li, H. Xu, S.-Q. Zang and T. C. W. Mak, Chem. Commun., 2015, 52, 525–528 RSC
- J.-J. Liu, Y. Wang, Y.-J. Hong, M.-J. Lin, C.-C. Huang and W.-X. Dai, Dalton Trans., 2014, 43, 17908–17911 RSC
- J. Z. Liao, X. Y. Wu, J. P. Yong, H. L. Zhang, W. B. Yang, R. Yu and C. Z. Lu, Cryst. Growth Des., 2015, 15, 4952–4958 CAS
- W.-Q. Fu, M. Liu, Z.-G. Gu, S.-M. Chen and J. Zhang, Cryst. Growth Des., 2016, 16, 5487–5492 CAS
- I. M. Walton, J. M. Cox, T. B. Mitchell, N. P. Bizier and J. B. Benedict, CrystEngComm, 2016, 18, 7972–7977 RSC
- X. Yu, Z. Wang, M. Buchholz, N. Füllgrabe, S. Grosjean, F. Bebensee, S. Bräse, C. Wöll and L. Heinke, Phys. Chem. Chem. Phys., 2015, 17, 22721–22725 RSC
- Z. Wang, L. Heinke, J. Jelic, M. Cakici, M. Dommaschk, R. J. Maurer, H. Oberhofer, S. Grosjean, R. Herges, S. Brase, K. Reuter and C. Woll, Phys. Chem. Chem. Phys., 2015, 17, 14582–14587 RSC
- X. Xing, Z. Chen, L. Cai, C. Sun, L. Cai, M. Wang and G. Guo, RSC Adv., 2016, 6, 24190–24194 RSC
- S. Hu, M. You, S. Chen and Z. Fu, CrystEngComm, 2016, 18, 7221–7224 RSC
- K. Healey, W. Liang, P. D. Southon, T. L. Church and D. M. D'Alessandro, J. Mater. Chem. A, 2016, 1–4 Search PubMed
- Y. X. Xie, W. N. Zhao, G. C. Li, P. F. Liu and L. Han, Inorg. Chem., 2016, 55, 549–551 CrossRef CAS PubMed
- J. Park, D. Feng, S. Yuan and H. C. Zhou, Angew. Chem., Int. Ed., 2015, 54, 430–435 CrossRef CAS PubMed
- C. Zhang, L. Sun, C. Zhang, S. Wan, Z. Liang and J. Li, Inorg. Chem. Front., 2016, 3, 814–820 RSC
- O. Toma, N. Mercier, M. Allain, A. A. Kassiba, J. P. Bellat, G. Weber and I. Bezverkhyy, Inorg. Chem., 2015, 54, 8923–8930 CrossRef CAS PubMed
- C. Zhang, L. Sun, Y. Yan, Y. Liu, Z. Liang, Y. Liu and J. Li, J. Mater. Chem. C, 2017, 5, 2084–2089 RSC
- J. Wang, S. L. Li and X. M. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 24862–24869 CAS
- H. Y. Li, Y. L. Wei, X. Y. Dong, S. Q. Zang and T. C. W. Mak, Chem. Mater., 2015, 27, 1327–1331 CrossRef CAS
- C. J. Zhang, Z. W. Chen, R. G. Lin, M. J. Zhang, P. X. Li, M. S. Wang and G. C. Guo, Inorg. Chem., 2014, 53, 847–851 CrossRef CAS PubMed
- G. Mehlana, S. A. Bourne and G. Ramon, Dalton Trans., 2012, 41, 4224–4231 RSC
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2011, 112, 1105–1125 CrossRef PubMed
- Y. Gong, Y. Zhou, J. Li, R. Cao, J. Qin and J. Li, Dalton Trans., 2010, 39, 9923–9928 RSC
- K. Uemura, S. Kitagawa, M. Kondo, K. Fukui, R. Kitaura, H.-C. Chang and T. Mizutani, Chem. – Eur. J., 2002, 8, 3586–3600 CrossRef CAS
- M. Du, X.-G. Wang, Z.-H. Zhang, L.-F. Tang and X.-J. Zhao, CrystEngComm, 2006, 8, 788–793 RSC
- D. Chisca, L. Croitor, O. Petuhov, E. B. Coropceanu and M. S. Fonari, CrystEngComm, 2016, 18, 38–41 RSC
- G. Park, H. Yang, T. H. Kim and J. Kim, Inorg. Chem., 2011, 50, 961–968 CrossRef CAS PubMed
- Z.-Z. Lu, R. Zhang, Y.-Z. Li, Z.-J. Guo and H.-G. Zheng, J. Am. Chem. Soc., 2011, 133, 4172–4174 CrossRef CAS PubMed
- A. Mallick, B. Garai, M. A. Addicoat, P. S. Petkov, T. Heine and R. Banerjee, Chem. Sci., 2015, 6, 1420–1425 RSC
- G. Mehlana, S. A. Bourne, G. Ramon and L. Öhrström, Cryst. Growth Des., 2013, 13, 633–644 CAS
- G. Mehlana, G. Ramon and S. A. Bourne, CrystEngComm, 2013, 15, 9521 RSC
- G. Mehlana, G. Ramon and S. A. Bourne, Microporous Mesoporous Mater., 2016, 231, 21–30 CrossRef CAS
- G. Mehlana, V. Chitsa and T. Mugadza, RSC Adv., 2015, 5, 88218–88233 RSC
- C.-Y. Sun, X.-L. Wang, C. Qin, J.-L. Jin, Z.-M. Su, P. Huang and K.-Z. Shao, Chem. – Eur. J., 2013, 19, 3639–3645 CrossRef CAS PubMed
- Z.-J. Lin, J. Lü, M. Hong and R. Cao, Chem. Soc. Rev., 2014, 43, 5867–5895 RSC
- F. Llabresixamena, O. Casanova, R. Galiassotailleur, H. Garcia and A. Corma, J. Catal., 2008, 255, 220–227 CrossRef CAS
- M. A. Rawashdeh-Omary, M. D. Rashdan, S. Dharanipathi, O. Elbjeirami, P. Ramesh and H. V. R. Dias, Chem. Commun., 2011, 47, 1160–1162 RSC
- Y. Yu, J. P. Ma, C. W. Zhao, J. Yang, X. M. Zhang, Q. K. Liu and Y. Bin Dong, Inorg. Chem., 2015, 54, 11590–11592 CrossRef CAS PubMed
- N.-N. Yang, W. Sun, F.-G. Xi, Q. Sui, L.-J. Chen and E.-Q. Gao, Chem. Commun., 2017, 53, 1747–1750 RSC
- C. G. Granqvist, Sol. Energy Mater. Sol. Cells, 2007, 91, 1529–1598 CrossRef CAS
- C. E. Sing, J. Kunzelman and C. Weder, J. Mater. Chem., 2009, 19, 104 RSC
- L. Liu, S. Peng, W. Wen and P. Sheng, Appl. Phys. Lett., 2007, 90, 0–3 Search PubMed
- J. Livage, Coord. Chem. Rev., 1999, 190, 391–403 CrossRef
- R. Gavara, C. A. T. Laia and A. Jorge Parola, Chem. – Eur. J., 2010, 16, 7760–7766 CrossRef CAS PubMed
- M. Zhou, J. Bao, M. Tao, R. Zhu, Y. Lin, X. Zhang and Y. Xie, Chem. Commun., 2013, 49, 6021–6023 RSC
- J. Schwiertz, A. Geist and M. Epple, Dalton Trans., 2009, 2921–2925 RSC
- G. Mehlana, C. Wilkinson, G. Ramon and S. Bourne, Polyhedron, 2015, 98, 224–229 CrossRef CAS
- S. Zhan, M. Li, W. Ng and D. Li, Chem. – Eur. J., 2013, 19, 10217–10225 CrossRef CAS PubMed
- H. Hara, A. Kobayashi, S. Noro, H.-C. Chang and M. Kato, Dalton Trans., 2011, 40, 8012–8018 RSC
- N. F. Sciortino, K. R. Scherl-Gruenwald, G. Chastanet, G. J. Halder, K. W. Chapman, J. F. L. Letard and C. J. Kepert, Angew. Chem., Int. Ed., 2012, 51, 10154–10158 CrossRef CAS PubMed
- P. Li, M. Wang, L. Cai, G. Wang and G. Guo, J. Mater. Chem. C, 2014, 3, 253–256 RSC
- D. Chisca, L. Croitor, E. B. Coropceanu, O. Petuhov, S. G. Baca, K. Krämer, S.-X. Liu, S. Decurtins, H. J. Rivera-Jacquez, A. E. Masunov and M. S. Fonari, CrystEngComm, 2016, 18, 384–389 RSC
- P.-C. Guo, T.-Y. Chen, X.-M. Ren, Z. Chu and W. Jin, J. Mater. Chem. A, 2014, 2, 13698–13704 CAS
- J.-P. Ma, S.-C. Liu, C.-W. Zhao, X.-M. Zhang, C.-Z. Sun and Y.-B. Dong, CrystEngComm, 2014, 16, 304–307 RSC
- D. Chisca, L. Croitor, E. B. Coropceanu, O. Petuhov, G. F. Volodina, S. G. Baca, K. Krämer, J. Hauser, S. Decurtins, S.-X. Liu and M. S. Fonari, Cryst. Growth Des., 2016, 16, 7011–7024 CAS
- A. Seeboth, D. Loetzsch and R. Ruhmann, American Journal of Materials Science, 2011, 1, 139–142 CrossRef
- H. D. Takagi, K. Noda, S. Itoh and S. Iwatsuki, Platinum Met. Rev., 2004, 48, 117–124 CrossRef CAS
- G.-G. Shan, H.-B. Li, H.-T. Cao, D.-X. Zhu, P. Li, Z.-M. Su and Y. Liao, Chem. Commun., 2012, 48, 2000 RSC
- M. Andrzejewski and A. Katrusiak, J. Phys. Chem. Lett., 2017, 8, 279–284 CrossRef CAS PubMed
- M. Andrzejewski and A. Katrusiak, J. Phys. Chem. Lett., 2017, 8, 929–935 CrossRef CAS PubMed
- A. Bacchi, S. Bourne, G. Cantoni, S. A. M. Cavallone, S. Mazza, G. Mehlana, P. Pelagatti and L. Righi, Cryst. Growth Des., 2015, 15, 1876–1888 CAS
- D. Dubbeldam, L. Bellarosa and N. Lo, J. Phys. Chem. C, 2013, 117, 20706–20714 Search PubMed
| This journal is © The Royal Society of Chemistry 2017 |