
Enzyme encapsulation in metal–organic frameworks for applications in catalysis
Marek B.
Majewski†
 ab,
Ashlee J.
Howarth†
a,
Peng
Li†
a,
Michael R.
Wasielewski
ab,
Joseph T.
Hupp
ab and
Omar K.
Farha
*ac
ab,
Ashlee J.
Howarth†
a,
Peng
Li†
a,
Michael R.
Wasielewski
ab,
Joseph T.
Hupp
ab and
Omar K.
Farha
*ac
aDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208-3113, USA. E-mail: o-farha@northwestern.edu
bArgonne-Northwestern Solar Energy Research (ANSER) Center, Northwestern University, Evanston, IL 60208-3113, USA
cDepartment of Chemistry, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia
First published on 20th February 2017
Abstract
Enzymes are natural catalysts which are highly selective and efficient. Given that enzymes have very intricate and delicate structures, they need to be stabilized and protected by a support material if they are to be used under challenging catalytic conditions. This highlight focuses on the use of metal–organic frameworks as supports for enzyme encapsulation and subsequent catalytic applications. De novo and post-synthetic methods of encapsulation are discussed and the relative catalytic activities of the enzyme@MOF composites versus free enzymes are highlighted.
 Marek B. Majewski | Marek B. Majewski is a postdoctoral fellow at the Argonne-Northwestern Solar Energy Research (ANSER) Center working with Prof. Wasielewski, Prof. Hupp and Prof. Farha. He completed his B.Sc. in 2007 at the University of Saskatchewan followed by his Ph.D. degree in 2013 under the mentorship of Prof. Michael Wolf at the University of British Columbia. Currently, his research revolves around photoinitiated charge separation and transport to drive molecular solar fuels catalysts and the design of functional inorganic materials. |
 Ashlee J. Howarth | Ashlee J. Howarth is an NSERC postdoctoral fellow in the department of Chemistry at Northwestern University working with Prof. Hupp and Prof. Farha. She received her B.Sc. degree in 2009 from The University of Western Ontario and went on to complete a Ph.D. degree in Chemistry at the University of British Columbia under the supervision of Prof. Michael Wolf. Her current research focuses primarily on wastewater remediation using metal–organic frameworks (MOFs) as adsorbents. |
 Peng Li | Peng Li was born in Hunan, China. He received his B.S. (2006) and M.S. (2009) degrees from Fudan University in China, and Ph.D. (2014) from University of Texas at San Antonio under the supervision of Prof. Banglin Chen. He joined Prof. Joseph T. Hupp and Prof. Omar K. Farha's Group in Northwestern University as a postdoctoral fellow since 2014. His research interest is related to functional porous materials for gas storage, separations, catalysis, and enzyme immobilization. |
 Michael R. Wasielewski | Michael R. Wasielewski is the Clare Hamilton Hall Professor of Chemistry at Northwestern University, Executive Director of the Institute for Sustainability and Energy at Northwestern (ISEN), and Director of the Argonne-Northwestern Solar Energy Research (ANSER) Center, a DOE Energy Frontier Research Center. He received his Ph.D. from the University of Chicago and was a postdoctoral fellow at Columbia University and Argonne National Laboratory. His research focuses on light-driven processes in molecules and materials, artificial photosynthesis, molecular electronics, and molecular spintronics. |
 Joseph T. Hupp | Joseph T. Hupp is a Morrison Professor of Chemistry at Northwestern University. He was a student of the late Mike Weaver at Michigan State University and Purdue University, completing a Ph.D. degree in 1983. He was a postdoc at the University of North Carolina. His research centres on energy- and defense-relevant materials chemistry, including chemical storage and separations, catalysis, light-to-electrical energy conversion and catalytic water oxidation. His research findings place him among the world's most highly cited chemists as assessed by Thomson-Reuters. |
 Omar K. Farha | Omar K. Farha is a research professor of chemistry at Northwestern University, Distinguished Adjunct Professor at King Abdulaziz University, Chief Scientific Officer of NuMat Technologies, and associate editor for ACS Applied Materials and interfaces. His current research spans diverse areas of chemistry and materials science ranging from energy to defense related challenges. Specifically, his research focuses on the rational design of metal–organic frameworks (MOFs) and porous-organic polymers for sensing, catalysis, storage, separations and light harvesting. Omar was named by Thomson Reuters one of the “Highly Cited Researchers” in 2014, 2015, and 2016. |
1. Introduction
Throughout our existence, humans have looked to Nature and natural systems for inspiration in solving complex problems such as self-healing,1 aerodynamics,2 solar energy harvesting3 and catalysis.4,5 Biological processes have been subject to millions of years of evolutionary experimentation giving rise to structures and functions with remarkable efficacy.6 One class of biomacromolecules of interest from a biomimetic7 standpoint is enzymes – Nature's catalysts.8,9 Enzymes are linear sequences of amino acids that fold to give intricate structures with highly specific catalytically active sites. As a result, enzymes produce highly selective (regio-, stereo-, chemo-) products with accelerated reaction rates and high turnover numbers.10 Given that enzymes are selective, efficient and environmentally benign catalysts, they have found application in a handful of large-scale processes such as the production of fine and pharmaceutical chemicals.11–13 Advances in protein engineering, sequence analysis and computational modelling allow for the fine tuning of enzymes to control aspects such as substrate recognition, efficiency and the nature of the product formed.8,9 The ability to make designer enzymes tailored for specific chemical transformations makes biocatalysis a very promising avenue for a variety of industrial processes.14 The application of enzymes thus far however, has been limited, in part due to the lack of long-term stability and difficulties with recyclability and recovery.15 The immobilization of enzymes on solid supports is one possible solution for promoting the industrialization of enzymes as catalysts.15,16 Immobilization can lead to increased enzyme stability, handling and recoverability-factors that in turn reduce cost. Solid supports that have been studied for enzyme immobilization include, but are not limited to, silicate glass,17,18 graphene oxide,19 carbon nanotubes,20 nanoparticles,21 mesoporous silica,22 macroporous polymeric beads such as Eupergit C® (ref. 23, 24) and metal–organic frameworks (MOFs).25–27Metal–organic frameworks offer many intriguing properties that make them of interest for enzyme immobilization.25–28 Given that MOFs are composed of both inorganic and organic components, several kinds of interactions between the MOF support and enzyme are possible. Among them are hydrogen bonding, including salt-bridge formation; other van der Waals forces, including dispersion forces; and covalent and/or coordinative bonding. Dispersion forces, while typically negligible for small molecules, can be large (and even predominant) for macromolecules, where their magnitude scales with the number of polarizable electrons. Since the strength of dispersion interactions varies strongly with distance, appropriate size matching of MOF pores to enzyme axes should enhance these interactions. The crystalline and ordered nature of MOFs allows for uniform loading, less leaching and an understanding of the local environment around the immobilized enzyme.25,29 The structures of MOFs are highly tunable such that surface area as well as pore size, shape and volume can be optimized for the immobilization and/or encapsulation of specific enzymes. Lastly, MOFs can be designed to be robust under harsh thermal, mechanical and chemical conditions30 which is important for immobilization and subsequent protection of enzymes to be used under challenging catalytic conditions. While enzymes can be immobilized on the surface of MOFs,31–39 this highlight will focus on examples of enzymes immobilized inside MOFs (i.e. encapsulation) with an emphasis on catalytic applications. The most obvious potential benefit of encapsulation is physical inhibition of enzyme denaturation (i.e. unfolding or changing of shape) in response, for example, to heating, dehydration, or changes in solution ionic strength. With good size matching between enzymes and MOF pores, encapsulation can also serve to prevent enzyme deactivation via aggregation. Although not yet widely explored, appropriately tailored MOF pores may provide local buffering or otherwise optimize the enzyme's microenvironment. The synthetic methods used to encapsulate enzymes and important design rules for MOF-based bioreactors will be discussed.
2. Enzyme immobilization in MOFs
One can imagine that biomacromolecules such as enzymes may be encapsulated within MOFs via two general strategies: by assembling the MOF around the enzyme (which we term de novo encapsulation) or by introducing the enzyme into the pre-existing MOF (which we term post-synthetic encapsulation). Each route manifests implications regarding preserving enzyme activity, efficiency and stability while at the same time dictating the conditions that may be used to prepare the supporting framework. In addition to considerations regarding the method of encapsulation, in systems prepared for catalytic applications, it is important to consider the effect that the chosen framework may have on factors such as substrate diffusion and/or selectivity. Coefficients for molecular diffusion through MOFs are typically much smaller than for diffusion through aqueous solutions. Thus, even MOF crystallite size can be important, as diffusion times increase as the square of the distance travelled. Clearly, in order to maximize catalyst performance, a delicate balance of these factors must be considered when selecting or tailoring supported catalytic systems for specific applications.We begin with examples of de novo encapsulation methods via processes known as coprecipitation and biomineralization, two related strategies each with its own advantages. Subsequently, we discuss post-synthetic methods of enzyme encapsulation using both channel- and cage-type MOFs as examples (Fig. 1).
 | ||
| Fig. 1 Methods used to encapsulate enzymes in MOFs. | ||
2.1 Enzyme incorporation de novo
In general, de novo approaches revolve around combining the organic and inorganic building blocks of the framework together with the target enzyme under conditions that favor both framework formation and preserve the tertiary structure of the active enzyme. Most MOFs present channels and/or apertures that are smaller than the size of the folded enzyme (see section 2.2) – thus the need to assemble the MOF around the enzyme. Underscoring the novelty of this burgeoning field, little (non-MOF) literature precedent exists for this approach; however, within the MOF field several groups have reported compelling examples of its applicability. | ||
| Fig. 2 Encapsulation of Cyt c in ZIF-8 by coprecipitation for the detection of organic peroxides. | ||
Although coprecipitation provides a direct and relatively facile route to the formation of these biocomposite materials, employing synthetic conditions that require the target enzyme to be independently stabilized against denaturation is a complicating factor. ZIF-8 is a framework with relatively small apertures (ca. 0.35 nm static diameter, dynamically expandable to about 0.5 nm). The consequences are slow substrate diffusion (compared to aqueous solution),41 thereby potentially limiting the catalytic activity of encapsulated enzymes, as well through exclusion of candidate substrates or co-reactants larger than ca. 0.5 nm. Nevertheless, the ZIF family of frameworks is a logical foundation for the coprecipitation method owing to the mild conditions that can be used for synthesis, the synthetic ease of linker modification to enhance framework-enzyme interactions, and the resistance of many ZIFs to degradation by water.41–43
To combat enzyme denaturation Shieh et al. pursued de novo enzyme encapsulation (catalase in ZIF-90; Fig. 3) in aqueous solution.44 Catalase (CAT), an industrially important enzyme employed for wastewater remediation, must be shielded in a matrix to prevent interaction with protease; an enzyme typically used up-stream of catalase. Although the peroxidase activity of catalase is retained, the report from Shieh, et al. illustrates some of the complications of de novo enzyme encapsulation by comparing composites formed in water versus ethanol. Perhaps unsurprisingly, using the conventional ethanol-based ZIF-90 preparation method yields a CAT@ZIF-90 composite that is catalytically inactive. In contrast, the aqueous sample is catalytically active, yielding an observed rate constant (kobs) of 0.0268 s−1. Further highlighting the degree of optimization complexity in these types of systems, Shieh and coworkers report that the rate constant with this composite is lower than that with free CAT in solution (kobs = 0.897 s−1), implying a non-ideal interface between ZIF-90 and catalase, some degree of catalase denaturation, and/or mass transport limitations.
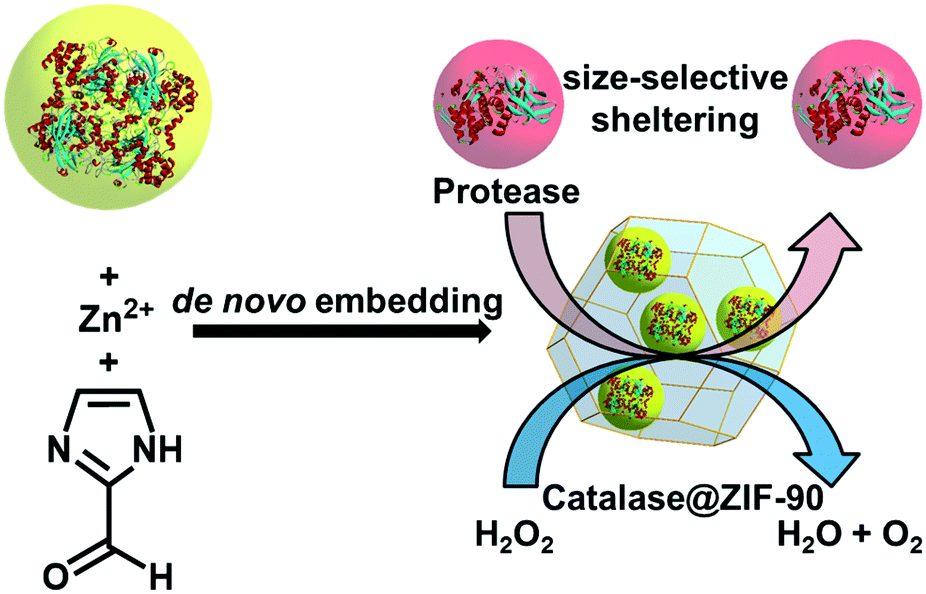 | ||
| Fig. 3 De novo encapsulation of catalase in ZIF-90. | ||
In an attempt to impart multifunctionality to these new materials, Wu and coworkers reported the successful encapsulation of two different enzymes, glucose oxidase (GOx) and horseradish peroxidase (HRP) in ZIF-8 via coprecipitation.45 Confocal laser scanning microscopy of composites containing fluorescein isothiocyanate (FITC) labelled GOx and HRP confirmed the presence of both enzymes within the crystal. In a display of intricate chemical cooperativity, catalytic efficiency of this composite was measured by indirectly detecting glucose through the formation of negatively charged 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS˙−) which has an electronic absorption band centered at 415 nm. In this scheme, GOx converts glucose into gluconic acid while concomitantly generating H2O2 that is then used as a substrate by HRP to oxidize ABTS2− to ABTS˙−. Notably, this method led to a fair detection limit of 0.5 μM. Denaturation and degradation of this pair of enzymes is thwarted by the rigid ZIF-8 framework, which protects the encapsulated biomacromolecules from digesting enzymes and from chelating compounds, as illustrated experimentally by exposure of the composite to dissolved ethylenediaminetetraacetic acid. As expected, this composite also exhibits improved thermal stability versus the enzyme in solution.
 | ||
| Fig. 4 Stability of horseradish peroxidase (HRP) encapsulated in ZIF-8 via biomineralization compared to other supports under varying conditions. Reproduced from ref. 46. | ||
A direct comparison of composite urease@ZIF-8 prepared by coprecipitation (carried out in the presence of PVP in aqueous solution) and by biomimetic mineralization yielded similar encapsulation efficiencies (as determined by the amount of FITC labelled urease incorporated into the final material). Notably, the coprecipitation method resulted in smaller crystal sizes (120 nm versus 500 nm) resulting in an increased rate of enzymatic reaction, while the composites prepared by biomineralization exhibited enhanced stability over a larger breadth of temperatures.47,48
Although viable as strategies for embedding biomacromolecules such as enzymes in MOFs, coprecipitation and biomimetic mineralization are also susceptible to complications. Namely, for both methods, reaction conditions must be chosen such that framework formation is possible while preserving the activity and integrity of the target enzyme. This narrows the scope of reaction media largely to aqueous solutions, a synthetic condition not common for MOF synthesis.49 Additionally, controlling biocomposite crystal size when employing these methodologies is challenging, resulting in materials that vary in catalytic activity.
One hybrid in situ encapsulation approach recently reported involves the formation of MIL-88A(Fe) hollow spheres via in-droplet microfluidics. These hollow spheres were used to encapsulate glycerol dehydrogenase, horseradish peroxidase and acetylcholinase.50 Conversely, another hybrid approach involves the formation of MOF particles around a pre-formed microcapsule where an emulsion containing Candida antartica lipase B (CALB) was stabilized with UiO-66 nanoparticles and the capsule's core was further stabilized against degradation via growth of a ZIF-8 shell.51 These methods, although more complicated than the strictly de novo enzyme encapsulation methods, allow for a greater degree of composite complexity while still preserving enzyme activity.
2.2 Post-synthetic enzyme incorporation
Enzymes can also be encapsulated in MOFs post-synthetically. This method typically involves soaking MOF crystals in a solution of the desired enzyme for anywhere from minutes to days at room or slightly elevated temperatures (e.g., 37 °C) depending on the system. The enzymes to be immobilized are often dissolved in water or a buffer solution at physiological pH (∼7.4). To identify the optimal solution for post-synthetic enzyme encapsulation, it is important not only to consider the pH at which both the enzyme and MOF are stable but also the chemical identity of the buffer components. For example, many MOFs are not stable in phosphate buffer solutions even though the pH of these buffers is in the optimal physiological range.30 Post-synthetic enzyme encapsulation for catalytic applications has been demonstrated using both cage-type and channel-type MOFs and examples of each will be discussed below.The first example of enzyme encapsulation in a cage-type MOF was presented by Ma and coworkers where microperoxidase-11 (MP-11, 3.3 × 1.7 × 1.1 nm) was encapsulated in a Tb-based MOF (Tb-mesoMOF) with cages 3.9 and 4.7 nm in diameter and apertures of 1.3 and 1.7 nm, respectively (Fig. 5).52 In this example the shortest axis of the enzyme is small enough to allow enzyme diffusion into both cages while leaving the 0.9 nm micropores throughout the framework unoccupied to permit subsequent infiltration of the composite by substrate molecules. The peroxidase activity (oxidation of 3,5-di-t-butyl-catechol) and subsequent stability of the encapsulated MP-11 enzyme was compared to the free enzyme in HEPES buffer solution and also to MP-11 immobilized on the mesoporous silica support, MCM-41. Although the initial rate of catalytic oxidation was found to be faster in the free enzyme (8.93 × 10−4 mM s−1) compared to that immobilized in Tb-mesoMOF (7.58 × 10−5 mM s−1), the conversion after 25 hours was higher with the MOF composite (48.7%) than with the free enzyme (12.3%). While the silica and MOF composites showed similar initial reaction rates, the MOF-based bioreactor was shown to withstand 6 cycles of catalysis with negligible effect on the initial reaction rate whereas the MCM-41 composite showed a 60% decrease in the initial rate between the first and second reaction cycle. The superior recyclability of the MOF composite material was attributed to strong interactions between the hydrophobic MOF cages and the enzyme53 compared to the weak interactions with MCM-41 which led to detectable enzyme leaching. An alternative explanation for the observation of leaching from MCM-41 versus an absence with Tb-mesoMOF is simply that that the apertures of Tb-mesoMOF are too small to permit escape of the enzyme (but those for MCM-41 are not). That enzyme leaching is not observed with Tb-mesoMOF points to the stability of the MOF crystal in the reaction environment and also to the absence of defects of sufficient size to permit enzyme escape.
 | ||
| Fig. 5 Encapsulation of MP-11 in Tb-mesoMOF. | ||
Ma and coworkers also immobilized myoglobin (Mb, 2.1 × 3.5 × 4.4 nm)54 and cytochrome c (Cyt c, 2.6 × 3.2 × 3.3 nm)55 in Tb-mesoMOF. In these examples the enzymes must undergo conformational changes to enter the cages of the MOF through the 1.3 and 1.7 nm apertures. In the case of Mb, the peroxidase activity of the enzyme was retained after encapsulation, albeit with a slower initial reaction rate (8.33 × 10−6 mM s−1) than that of the free enzyme (3.27 × 10−4 mM s−1). The Mb@Tb-mesoMOF was shown to retain catalytic activity over 15 cycles however, compared to Mb immobilized on the mesoporous silica support, SBA-15, which showed a 40% decrease in reaction rate after the first cycle. It is important to note that the size of the unoccupied channels (0.9 nm) in the enzyme@Tb-mesoMOF composites dictates the size of the substrate that can access the enzyme. For example, no peroxidase activity was observed using Mb@Tb-mesoMOF with 2,2′-azinobis(3-ethyl-benzthiazoline)-6-sulfonate (ABTS, 1.0 × 1.7 nm) as a substrate whereas oxidation of 1,2,3-trihydroxybenzene (THB, 0.57 × 0.58 nm) was observed.54 While not discussed in the Mb@Tb-mesoMOF work, the channel size should also be exploitable to prevent inhibitors or denaturants from accessing the enzyme.
The design and construction of cage-type MOFs with larger pores was undertaken by Zhou et al. to give MOFs with fewer size limitations for enzyme encapsulation.56 PCN-333 has cages of 1.1, 3.4 and 5.5 nm in diameter with apertures of 2.6 and 3.0 nm in size. It was found that when encapsulating larger enzymes in PCN-333(Al) such as horseradish peroxidase (HRP, 4.0 × 4.4 nm × 6.8 nm) and Cyt c, the loading corresponds to encapsulation of 1 enzyme per cage, whereas with smaller enzymes such as MP-11, the loading corresponds to multiple enzymes incorporated per cage. In each case, the encapsulated enzyme demonstrates higher stability compared to the free enzyme, but lower initial reaction rates for oxidation of o-phenylenediamine (HRP) or ABTS (Cyt c, MP-11). The low initial reaction rates may be attributed to restricted diffusion of reactants and products and/or low enzyme accessibility in the cage-type structure. In order to overcome problems with reactant and product diffusion, Zhou and coworkers constructed PCN-888, an Al-MOF with cages of 2.0, 5.0 and 6.2 nm in diameter and apertures of 2.5 and 3.6 nm in size.57 PCN-888 was used to selectively encapsulate glucose oxidase (GOx, 6.0 × 5.2 × 7.7 nm) in the largest cage and HRP in the medium sized cage to give a tandem bioreactor with the smallest cages being unoccupied by enzymes, and therefore available for reactant and product transport (Fig. 6). In addition, the encapsulated enzymes were shown to be more stable than the free enzymes at slightly elevated temperature (37 °C) in the presence of trypsin. Although the initial reaction rate for the encapsulated enzyme is much closer to that of the free enzyme (compared to other examples of enzymes encapsulated in cage-type MOFs), the reaction rate is still slower than that found with the free enzyme.
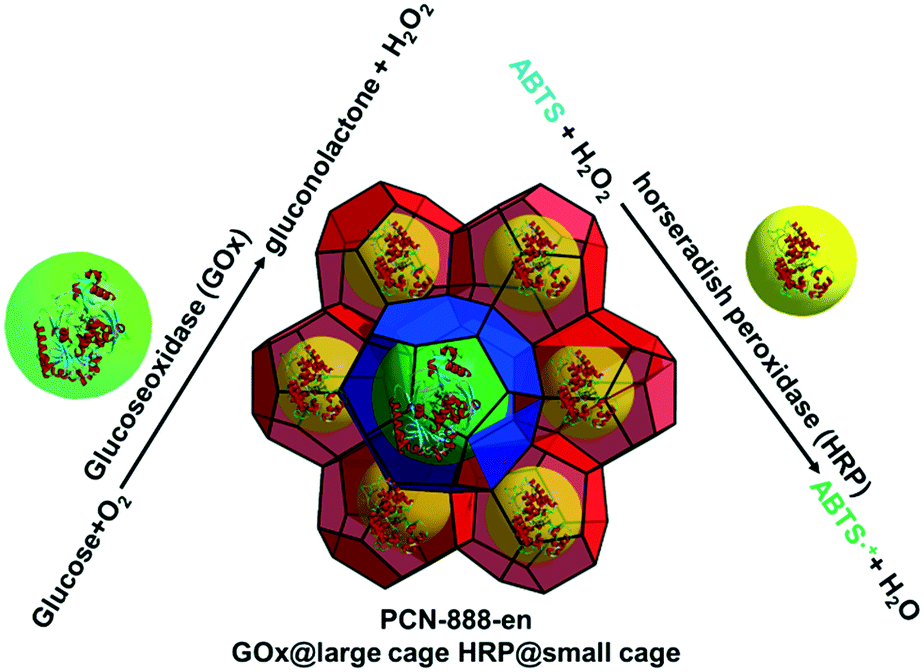 | ||
| Fig. 6 Encapsulation of GOx and HRP in PCN-888 for tandem catalysis. | ||
 | ||
| Fig. 7 Encapsulation of GFP in IRMOF-74-IX. | ||
The ideal MOF-based bioreactor should encapsulate and protect the desired enzyme but also allow for relatively unimpeded diffusive transport of reactants and products to and from the encapsulated enzyme, even when the enzyme is sited deep within the MOF. Hierarchical channel-type MOFs with windows between the channels are promising in this regard since large channels can be designed to fit the desired enzyme while small channels can remain unoccupied by enzymes and thus available for diffusive transport of small molecules. Windows between the channels can allow reactants access to the encapsulated enzyme in the larger neighbouring channels. We first demonstrated this concept using the hierarchical MOF, NU-1000, and the enzyme, cutinase.60
NU-1000 has triangular channels with an edge length of 0.9 nm and hexagonal channels which are 3.1 nm in diameter61 meaning that cutinase (smallest axis is 3.0 nm) can be encapsulated in the larger pores while leaving the smaller pores open for delivery of reactants and release of products. Although cutinase loading in NU-1000 is not as high as in PCN-600 (a MOF with channels of only one size), experiments showed that more than 90% of the enzymes loaded in NU-1000 are accessible while only 6% of those in PCN-600 can be accessed. These findings highlight the importance of a hierarchical structure where smaller, enzyme-free channels enable molecular species, including solvent, to access the MOF interior, while windows between the smaller and larger channels permit molecules to reach enzymes throughout the MOF.
Using the same strategy, organophosphorus acid anhydrolase (OPAA, small axis is 4.4 nm) was encapsulated in PCN-128y, a MOF with hierarchical structure and larger hexagonal channels (4.4 nm) than NU-1000 to accommodate the larger enzyme.62 The encapsulated OPAA enzyme was found to be more stable than the free enzyme, retain catalytic activity at temperatures up to 70 °C and after being dried and stored at room temperature for 3 days. Although the MOF was successful in stabilizing the OPAA enzyme, the initial rate of reaction for the hydrolysis of soman (56–75 μmol min−1 mg−1) was not as high as that found using the free enzyme (305 μmol min−1 mg−1). To enhance the reaction rate, NU-1003, a MOF with hierarchical structure containing 4.4 nm hexagonal channels, 1.7 nm triangular channels and windows between the channels of 1.2 × 1.3 nm was designed to help promote molecular (reactant, product, solvent) transport and enzyme accessibility (Fig. 8).63 In addition, the size of NU-1003 was controlled to give average MOF crystallites as particles ranging from 10 μm down to 300 nm to further enhance diffusion. By capitalizing on these design concepts, we obtained a thus far rare example of a MOF-enzyme composite for which the initial rate of the catalyzed reaction is faster than that obtained with the free enzyme. Thus, the encapsulated enzyme OPAA was not only protected but also showed an initial reaction rate of 960 μmol min−1 mg−1 for soman hydrolysis that is more than 3 times faster than that of the free enzyme (i.e., detergent-stabilized enzyme in buffered aqueous solution).
 | ||
| Fig. 8 Representation of encapsulation of organophosphorus acid anhydrolase (OPAA) in the hierarchical channel-type Zr-MOF, NU-1003. | ||
3. Summary and outlook
As supports for stabilizing enzymes against environmental and incidental degradation, MOFs are an attractive target owing to their well-documented chemical and thermal stability. In addition, the immense library of known frameworks and the characterization of their respective properties and inherent stabilities paired with high porosity lends this class of materials to the formation of enzyme@MOF composites. Stabilizing enzymes against denaturation, while preserving native conformations of the biomacromolecule becomes especially important when encapsulating enzymes for specific catalytic purposes. The examples outlined here illustrate that enzyme@MOF composites can be effective at preserving enzyme activity while enforcing a greater degree of stability under catalytically relevant, but distinctly abiotic, conditions. Apart from a few examples, initial reaction rates for enzyme@MOF composites are not competitive with those of the free enzymes – but much has been learned about different encapsulation strategies and some design rules now exist for preparing enzyme@MOF composites that not only protect the encapsulated enzyme but also promote diffusion of reactants and products. It should be noted that the examples discussed throughout this highlight are based on the findings presented by different authors – in some cases the low initial reaction rates for enzyme@MOF composites compared to free enzymes may be a result of reactants/products not fitting through MOF apertures. In these instances, only enzymes immobilized on the surface of the MOFs may be active while all enzymes loaded into the framework may be assumed to be active. Thus, it is important to consider not only the size of the enzymes being encapsulated but also the size of the reactants/products of the reaction of interest.Current research efforts into enzyme@MOF composites that were not discussed in this highlight include encapsulating enzymes into materials for chemical sensing.64–69 One can imagine designing hybrid composites, where complex systems that first initiate a catalytic transformation are combined with a sensing component that detects specific catalytic products leading to direct measures of catalytic efficiency, reaction progress, or substrate concentration. One additionally important direction for the study of composites prepared from cage- and channel-type MOFs includes encapsulating enzymes into MOFs that feature high densities of missing-linker and/or missing-node type defects. This would allow for enzyme@MOF composites that were previously only attainable via de novo routes to be attainable post-synthetically.70
Significant progress has been made since the first example of enzyme encapsulation in a MOF was reported a decade ago. Hypothesis-driven research has led to enzyme@MOF composites that display initial catalytic reaction rates higher than those engendered by free enzymes. By using the set of design rules now established, together with others that await discovery, an expansive library of new functional composites should be designable and experimentally obtainable, perhaps contributing to the next wave of applied biocatalysis.
Acknowledgements
For work done in our own lab, we gratefully acknowledge the US Defense Threat Reduction Agency and the Army Research Office (grant HDTRA1-14-1-0014 and project W911NF-13-1-0229). This work was supported in part by the Argonne-Northwestern Solar Energy Research (ANSER) Center, an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, under award number DE-SC0001059 (M. B. M. for writing, M. R. W. and J. T. H. for editing). A. J. H. thanks NSERC for a postdoctoral fellowship.Notes and references
- C. E. Diesendruck, N. R. Sottos, J. S. Moore and S. R. White, Angew. Chem., Int. Ed., 2015, 54, 10428–10447 CrossRef CAS PubMed
.
- G. J. Parker, J. Mater. Sci.: Mater. Electron., 2010, 21, 965–979 CrossRef CAS
- P. O. Saboe, E. Conte, S. Chan, H. Feroz, B. Ferlez, M. Farell, M. F. Poyton, I. T. Sines, H. Yan, G. C. Bazan, J. Golbeck and M. Kumar, J. Mater. Chem. A, 2016, 4, 15457–15463 CAS
- X. Ma, A. C. Hortelão, T. Patiño and S. Sánchez, ACS Nano, 2016, 10, 9111–9122 CrossRef CAS PubMed
- C. E. Valdez, Q. A. Smith, M. R. Nechay and A. N. Alexandrova, Acc. Chem. Res., 2014, 47, 3110–3117 CrossRef CAS PubMed
-
T. W. Hanks and G. F. Swiegers, in Bioinspiration and Biomimicry in Chemistry, John Wiley & Sons, Inc., 2012, pp. 1–15 Search PubMed
- In this context we use “biomimetic” to mean deploying biocatalysts for abiotic reactions in abiotic environments.
- S. J. Benkovic and S. Hammes-Schiffer, Science, 2003, 301, 1196–1202 CrossRef CAS PubMed
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed
- S. F. M. van Dongen, J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, Angew. Chem., Int. Ed., 2014, 53, 11420–11428 CrossRef CAS PubMed
- H. Griengl, H. Schwab and M. Fechter, Trends Biotechnol., 2000, 18, 252–256 CrossRef CAS
- G. Hills, Eur. J. Lipid Sci. Technol., 2003, 105, 601–607 CrossRef CAS
- T. Nagasawa, T. Nakamura and H. Yamada, Appl. Microbiol. Biotechnol., 1990, 34, 322–324 CAS
- J.-M. Choi, S.-S. Han and H.-S. Kim, Biotechnol. Adv., 2015, 33, 1443–1454 CrossRef CAS PubMed
- R. DiCosimo, J. McAuliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437–6474 RSC
- M. C. R. Franssen, P. Steunenberg, E. L. Scott, H. Zuilhof and J. P. M. Sanders, Chem. Soc. Rev., 2013, 42, 6491–6533 RSC
- A. Küchler, J. N. Bleich, B. Sebastian, P. S. Dittrich and P. Walde, ACS Appl. Mater. Interfaces, 2015, 7, 25970–25980 Search PubMed
- S. Fornera, T. Bauer, A. D. Schluter and P. Walde, J. Mater. Chem., 2012, 22, 502–511 RSC
- I. V. Pavlidis, M. Patila, U. T. Bornscheuer, D. Gournis and H. Stamatis, Trends Biotechnol., 2014, 32, 312–320 CrossRef CAS PubMed
- W. Feng and P. Ji, Biotechnol. Adv., 2011, 29, 889–895 CrossRef CAS PubMed
- H. Jia, G. Zhu and P. Wang, Biotechnol. Bioeng., 2003, 84, 406–414 CrossRef CAS PubMed
- E. Magner, Chem. Soc. Rev., 2013, 42, 6213–6222 RSC
- E. Katchalski-Katzir and D. M. Kraemer, J. Mol. Catal. B: Enzym., 2000, 10, 157–176 CrossRef CAS
- P. Torres-Salas, A. del Monte-Martinez, B. Cutiño-Avila, B. Rodriguez-Colinas, M. Alcalde, A. O. Ballesteros and F. J. Plou, Adv. Mater., 2011, 23, 5275–5282 CrossRef CAS PubMed
- J. Mehta, N. Bhardwaj, S. K. Bhardwaj, K.-H. Kim and A. Deep, Coord. Chem. Rev., 2016, 322, 30–40 CrossRef CAS
- D. S. Raja, W.-L. Liu, H.-Y. Huang and C.-H. Lin, Comments Inorg. Chem., 2015, 35, 331–349 CrossRef
- X. Wu, M. Hou and J. Ge, Catal. Sci. Technol., 2015, 5, 5077–5085 CAS
- M. V. de Ruiter, R. Mejia-Ariza, J. J. L. M. Cornelissen and J. Huskens, Chem, 2016, 1, 29–31 CAS
- M. Hartmann, Chem. Mater., 2005, 17, 4577–4593 CrossRef CAS
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS
- W. Ma, Q. Jiang, P. Yu, L. Yang and L. Mao, Anal. Chem., 2013, 85, 7550–7557 CrossRef CAS PubMed
- F.-X. Qin, S.-Y. Jia, F.-F. Wang, S.-H. Wu, J. Song and Y. Liu, Catal. Sci. Technol., 2013, 3, 2761 CAS
- Y.-H. Shih, S.-H. Lo, N.-S. Yang, B. Singco, Y.-J. Cheng, C.-Y. Wu, I. H. Chang, H.-Y. Huang and C.-H. Lin, ChemPlusChem, 2012, 77, 982–986 CrossRef CAS
- C. Tudisco, G. Zolubas, B. Seoane, H. Zafarani, M. Kazemzad, J. Gascon, P. L. Hagedoorn and L. Rassaei, RSC Adv., 2016, 6, 108051–108055 RSC
- X. Wang, T. A. Makal and H.-C. Zhou, Aust. J. Chem., 2014, 67, 1629 CrossRef CAS
- W.-L. Liu, N.-S. Yang, Y.-T. Chen, S. Lirio, C.-Y. Wu, C.-H. Lin and H.-Y. Huang, Chem. – Eur. J., 2015, 21, 115–119 CrossRef CAS PubMed
- S. Patra, S. Sene, C. Mousty, C. Serre, A. Chaussé, L. Legrand and N. Steunou, ACS Appl. Mater. Interfaces, 2016, 8, 20012–20022 CAS
- L. Wen, A. Gao, Y. Cao, F. Svec, T. Tan and Y. Lv, Macromol. Rapid Commun., 2016, 37, 551–557 CrossRef CAS PubMed
- M. Zhao, X. Zhang and C. Deng, Chem. Commun., 2015, 51, 8116–8119 RSC
- F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761–5765 CrossRef CAS PubMed
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef CAS PubMed
- P. Chulkaivalsucharit, X. Wu and J. Ge, RSC Adv., 2015, 5, 101293–101296 RSC
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed
- F.-K. Shieh, S.-C. Wang, C.-I. Yen, C.-C. Wu, S. Dutta, L.-Y. Chou, J. V. Morabito, P. Hu, M.-H. Hsu, K. C. W. Wu and C.-K. Tsung, J. Am. Chem. Soc., 2015, 137, 4276–4279 CrossRef CAS PubMed
- X. Wu, J. Ge, C. Yang, M. Hou and Z. Liu, Chem. Commun., 2015, 51, 13408–13411 RSC
- K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 2015, 6, 7240 CrossRef CAS PubMed
- K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 2015, 6, 7240 CrossRef CAS PubMed
- K. Liang, C. J. Coghlan, S. G. Bell, C. Doonan and P. Falcaro, Chem. Commun., 2016, 52, 473–476 RSC
- Z. Hu, I. Castano, S. Wang, Y. Wang, Y. Peng, Y. Qian, C. Chi, X. Wang and D. Zhao, Cryst. Growth Des., 2016, 16, 2295–2301 CAS
- G.-Y. Jeong, R. Ricco, K. Liang, J. Ludwig, J.-O. Kim, P. Falcaro and D.-P. Kim, Chem. Mater., 2015, 27, 7903–7909 CrossRef CAS
- J. Huo, J. Aguilera-Sigalat, S. El-Hankari and D. Bradshaw, Chem. Sci., 2015, 6, 1938–1943 RSC
- V. Lykourinou, Y. Chen, X. S. Wang, L. Meng, T. Hoang, L. J. Ming, R. L. Musselman and S. Ma, J. Am. Chem. Soc., 2011, 133, 10382–10385 CrossRef CAS PubMed
- Y. Chen, S. Han, X. Li, Z. Zhang and S. Ma, Inorg. Chem., 2014, 53, 10006–10008 CrossRef CAS PubMed
- Y. Chen, V. Lykourinou, T. Hoang, L. J. Ming and S. Ma, Inorg. Chem., 2012, 51, 9156–9158 CrossRef CAS PubMed
- Y. Chen, V. Lykourinou, C. Vetromile, T. Hoang, L. J. Ming, R. W. Larsen and S. Ma, J. Am. Chem. Soc., 2012, 134, 13188–13191 CrossRef CAS PubMed
- D. Feng, T. F. Liu, J. Su, M. Bosch, Z. Wei, W. Wan, D. Yuan, Y. P. Chen, X. Wang, K. Wang, X. Lian, Z. Y. Gu, J. Park, X. Zou and H. C. Zhou, Nat. Commun., 2015, 6, 5979 CrossRef PubMed
- X. Lian, Y.-P. Chen, T.-F. Liu and H.-C. Zhou, Chem. Sci., 2016, 7, 6969–6973 RSC
- T. J. Pisklak, M. Macías, D. H. Coutinho, R. S. Huang and K. J. Balkus, Top. Catal., 2006, 38, 269–278 CrossRef CAS
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gandara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS PubMed
- P. Li, J. A. Modica, A. J. Howarth, E. L. Vargas, P. Z. Moghadam, R. Q. Snurr, M. Mrksich, J. T. Hupp and O. K. Farha, Chem, 2016, 1, 154–169 CAS
- J. E. Mondloch, W. Bury, D. Fairen-Jimenez, S. Kwon, E. J. DeMarco, M. H. Weston, A. A. Sarjeant, S. T. Nguyen, P. C. Stair, R. Q. Snurr, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 10294–10297 CrossRef CAS PubMed
- P. Li, S. Y. Moon, M. A. Guelta, S. P. Harvey, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2016, 138, 8052–8055 CrossRef CAS PubMed
- P. Li, S.-Y. Moon, M. A. Guelta, L. Lin, D. A. Gómez-Gualdrón, R. Q. Snurr, S. P. Harvey, J. T. Hupp and O. K. Farha, ACS Nano, 2016, 10, 9174–9182 CrossRef CAS PubMed
- C. M. Doherty, G. Grenci, R. Riccò, J. I. Mardel, J. Reboul, S. Furukawa, S. Kitagawa, A. J. Hill and P. Falcaro, Adv. Mater., 2013, 25, 4701–4705 CrossRef CAS PubMed
- C. Hou, Y. Wang, Q. Ding, L. Jiang, M. Li, W. Zhu, D. Pan, H. Zhu and M. Liu, Nanoscale, 2015, 7, 18770–18779 RSC
- X. Lu, X. Wang, L. Wu, L. Wu, Dhanjai, L. Fu, Y. Gao and J. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 16533–16539 CAS
- S. Patra, T. Hidalgo Crespo, A. Permyakova, C. Sicard, C. Serre, A. Chausse, N. Steunou and L. Legrand, J. Mater. Chem. B, 2015, 3, 8983–8992 RSC
- X. Wang, X. Lu, L. Wu and J. Chen, Biosens. Bioelectron., 2015, 65, 295–301 CrossRef CAS PubMed
- Y. Wang, C. Hou, Y. Zhang, F. He, M. Liu and X. Li, J. Mater. Chem. B, 2016, 4, 3695–3702 RSC
- Y. Kim, T. Yang, G. Yun, M. B. Ghasemian, J. Koo, E. Lee, S. J. Cho and K. Kim, Angew. Chem., Int. Ed., 2015, 54, 13273–13278 CrossRef CAS PubMed
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |