
General synthesis of metal oxide hollow core–shell microspheres as anode materials for lithium-ion batteries and as adsorbents for wastewater treatment†
Yuanxiang
Gu
 ,
Haolin
Wang
,
Yuxue
Xuan
,
Lei
Wang
* and
Yi
Qian
,
Haolin
Wang
,
Yuxue
Xuan
,
Lei
Wang
* and
Yi
Qian
Qingdao University of Science and Technology, Qingdao 266042, PR China. E-mail: inorchemwl@126.com
First published on 31st January 2017
Abstract
Hollow core–shell structures have many advantages, but their synthesis remains challenging. In this report, we demonstrate a general method for the preparation of a series of metal oxide hollow core–shell microspheres, including Co3O4, NiO and Mn2O3. The hollow core–shell microspheres were prepared via a coordination precipitation route followed by a calcination process. The morphology and microstructures of the metal oxide hollow core–shell microspheres could be controlled by optimizing the experimental conditions. A possible formation mechanism of the hollow core–shell structure was proposed. As anode materials for lithium ion cells, the Co3O4 hollow core–shell microspheres exhibited superior Li+ storage properties. When applied as adsorbents in water treatment, the NiO hollow core–shell microspheres showed an excellent ability to remove organic pollutants.
1. Introduction
In recent years, hollow core–shell structures are of great interest because of their outstanding properties such as low density, high surface area, and excellent surface permeability.1,2 Potential applications were found including lithium ion batteries, solar cells, drug delivery, water treatment, and sensors,3–7 which greatly inspired researchers to pay a lot of attention to designing and constructing hollow or core–shell micro- or nanostructures.8–11 The most popular approach for producing hollow core–shell structures is the template method, which can be simply classified into hard template and soft template methods.12–14 In particular, the templating approach utilizing carbonaceous microspheres as templates displays generality and availability, and has been used to successfully synthesize a number of multi-shelled hollow spheres of metal oxides.15,16 Although template-assisted methods have been proven to be very effective and versatile for the synthesis of hollow structures, they have many disadvantages. Specifically, not only is the fabrication process complicated, but special care must also be taken to prevent shell collapse during the template removal process. Recently, some chemical and physicochemical methods have also been developed to synthesize hollow core–shell structures, such as spray drying,17 interior corrosion,18 Ostwald ripening19 and the nanoscale Kirkendall effect.20 However, the above-mentioned methods are usually only suitable for certain specific metal oxide hollow structures, and strict synthetic conditions are required in most of the cases because the synthesis of hollow metal oxide core–shell structures is not only determined by their intrinsic structure but also significantly influenced by a series of external parameters, such as reaction time and temperature, the pH value of the precursor solution, and organic additives.21–23 Therefore, the preparation of scalable metal oxide microspheres with a hollow core–shell structure in a general and simple way remains a great challenge. Thus, the development of a novel and facile synthesis approach to prepare metal oxide hollow core–shell microspheres on a large scale would be of significant importance.In this report, hollow core–shell metal oxide microspheres including Co3O4, NiO and Mn2O3 were synthesized by a precipitation method combined with calcination. To the best of our knowledge, this is the first report on the synthesis of different metal oxide microspheres with a hollow core–shell structure by a general precipitation method without a template. The current approach to synthesize metal oxide hollow core–shell microspheres has special advantages including being ultra-facile without complicated procedures, general applicability in synthesizing metal oxide hollow core–shell microspheres, and the use of common chemical reagents without special reagents. These features are favorable for the scalable synthesis of metal oxide hollow core–shell microspheres for industrial applications. Herein, the effects of the reaction parameters on the morphology and microstructure of the materials, and their formation mechanism were discussed. As anode materials for lithium ion batteries, the electrochemical properties of the Co3O4 hollow core–shell microspheres were investigated, showing outstanding properties. Furthermore, the NiO hollow core–shell microspheres were used as adsorbents for wastewater treatment and showed an excellent ability to remove organic pollutants.
2. Experimental section
2.1. Synthesis of Co3O4, NiO and Mn2O3 hollow core–shell microspheres
All reagents were of analytical grade and used without further purification. In a typical experiment, 1.000 g of Co(Ac)2·4H2O (or Ni(Ac)2·4H2O) and 1.050 g of citric acid were dissolved in 15 mL of deionized water and 15 mL of dimethylformamide (DMF) under vigorous stirring in a beaker to form a homogeneous solution. Then, 10 mL of polyethylene glycol 400 (PEG-400) was introduced into the above solution under continuous stirring at room temperature. For the synthesis of hollow core–shell Mn2O3 microspheres, 1.260 g of Mn(Ac)2·4H2O and 1.260 g of citric acid were dissolved in 15 mL of deionized water and 15 mL of ethylene glycol (EG) under vigorous stirring to form a homogeneous solution. Then, 10 mL of PEG-200 was added to the above solution under continuous stirring. Subsequently, the above-obtained three solutions were all heated to 30 °C using a water bath. Along with the evaporation of solvents, precipitates were constantly produced. After 48 h, the precipitates were collected, centrifuged, washed with ethanol several times and dried at 80 °C for 10 h. Finally, the products were heated at a rate of 2 °C min−1 from room temperature to 500 °C, and kept at 500 °C for 4 h in a tube furnace in air.2.2. Characterization
The size and morphology of the samples were characterized using a field emission scanning electron microscope equipped with an energy-dispersive spectrometer (EDS) (FE-SEM, Hitachi S-4800, Japan). Transmission electron microscopy (TEM) images were taken using a Hitachi H-800 transmission electron microscope with an operating voltage of 100 kV. X-ray diffractometry (XRD) with a graphite monochromator and Cu Kα radiation (λ = 0.15148 nm) was applied to identify the phase of the samples at a step size of 0.02° while the voltage and electric current were held at 40 kV and 20 mA. The data were measured in the range of 10–70°. N2 adsorption–desorption isotherms were characterized on a Quadrasorb SI apparatus. The surface area was determined by the Brunauer–Emmett–Teller (BET) method, while the pore volume and pore-size distribution were determined via the nonlocal density functional theory (DFT) method. Thermal analysis (TG/DSC, DT-50 Setaram, France) was performed in an air atmosphere from 40 °C to 600 °C at a heating rate of 20 °C min−1. Infrared (IR) spectra were recorded with a FTIR spectrometer (Shimadzu) in the form of KBr pellets in the range of 400–4000 cm−1 with a spectral resolution of 2 cm−1.2.3. Electrochemical test
The electrodes were prepared by coating a slurry of the active material powders, acetylene black, and poly(vinylidene fluoride) binder (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:10 weight ratio) dissolved in N-methyl-2-pyrrolidinone onto a Cu foil substrate. The electrodes were dried in a vacuum at 100 °C for 24 h, and then assembled into a half-battery (CR2032 coin type) using lithium foil as a counter electrode in an Ar-filled glove box. The electrolyte used was 1 M LiPF6 in a mixed solvent containing ethylene carbonate and dimethyl carbonate in a 1:1 weight ratio. The charge–discharge characteristics of the cells were recorded on a LAND cell-testing system.
:20:10 weight ratio) dissolved in N-methyl-2-pyrrolidinone onto a Cu foil substrate. The electrodes were dried in a vacuum at 100 °C for 24 h, and then assembled into a half-battery (CR2032 coin type) using lithium foil as a counter electrode in an Ar-filled glove box. The electrolyte used was 1 M LiPF6 in a mixed solvent containing ethylene carbonate and dimethyl carbonate in a 1:1 weight ratio. The charge–discharge characteristics of the cells were recorded on a LAND cell-testing system.
2.4. Adsorption measurements
The adsorption properties were examined using Congo red. 0.02 mg of NiO microspheres was dispersed into 50 mL of Congo red aqueous solutions with different concentrations (10–300 mg L−1) under constant stirring at room temperature. After centrifugation, the adsorption phenomenon was investigated through the UV-vis spectrum of the supernatant. The concentration of Congo red was analyzed using a UV-vis spectrometer (Perkin-Elmer, Lambda-35). The characteristic absorption of Congo red at 500 nm was chosen as the monitored parameter for the adsorption process.3. Results and discussion
3.1. Morphology and microstructure of metal oxide hollow core–shell microspheres
The morphology and microstructure of the metal oxide hollow core–shell microspheres were characterized by SEM and TEM. The panoramic SEM image in Fig. 1a gives a representative overview of the Co3O4 sample after calcination at 500 °C. It can be seen that the calcined Co3O4 product exhibits a spherical morphology and has a mean diameter distribution of ca. 3.0 μm. As shown in Fig. 1b and its inset, it can be seen that the synthesized Co3O4 microspheres are made up of small particles with sizes of 20–40 nm and the surface of the microspheres has a porous structure. To investigate the interior structures, the microspheres were firmly ground and thus were further broken and collapsed. The close-up image of a single broken sphere (inset in Fig. 1a) shows that the Co3O4 microsphere has a core–shell structure. The TEM image (Fig. 1c) further confirms that the Co3O4 microspheres are characterized by hollow core–shell structures with a sphere-in-sphere morphology. Our synthesis method is very general. Besides Co3O4, this ultra-facile approach has also been extended to the synthesis of other metal oxide microspheres, including NiO and Mn2O3. Their corresponding SEM and TEM images are given in Fig. 1d–i. The NiO and Mn2O3 samples also exhibit spherical structures made up of small particles. Their corresponding TEM images shown in Fig. 1f and i confirm that the NiO and Mn2O3 microspheres also have hollow core–shell structures with a sphere-in-sphere morphology. The nitrogen adsorption–desorption isotherms and the corresponding pore size distribution curves of the Co3O4, NiO and Mn2O3 hollow core–shell microspheres are shown in Fig. S1.† The BET surface area value for the Co3O4, NiO and Mn2O3 hollow core–shell microspheres is calculated to be 21.06 m2 g−1, 20.51 m2 g−1 and 20.91 m2 g−1, respectively. Their average pore size is about 26 nm, 13 nm and 17 nm, respectively. The hollow core–shell structure with an inner surface can provide more active sites for redox reactions and the porous structure provides a short distance for ion diffusion, both being beneficial to the lithium storage capacity and adsorption capacity of the metal oxide microspheres. | ||
| Fig. 1 SEM and TEM images of metal oxide microspheres after calcination at 500 °C: Co3O4 (a–c), NiO (d–f) and Mn2O3 (g–i). Insets are the corresponding high-magnification SEM images of the Co3O4, NiO and Mn2O3 microspheres. | ||
In order to investigate the phase and structure of the obtained Co3O4 product, its corresponding XRD pattern is given in Fig. 2. All diffraction peaks of the Co3O4 product can be easily indexed to the pure cubic structure of Co3O4, which are in good agreement with the literature values (JCPDS 43-1003). The narrow sharp peaks in the XRD pattern indicate that the Co3O4 product is well crystallized. Furthermore, energy-dispersive spectrometry (EDS) was also utilized to identify the chemical element composition of the product. As shown in Fig. 2b, the calcined sample mainly contains Co and O elements, further confirming the formation of pure Co3O4. The XRD patterns of the NiO and Mn2O3 microspheres are given in Fig. S2.† Their corresponding XRD patterns also indicate the formation of pure NiO and Mn2O3 with good crystallization.
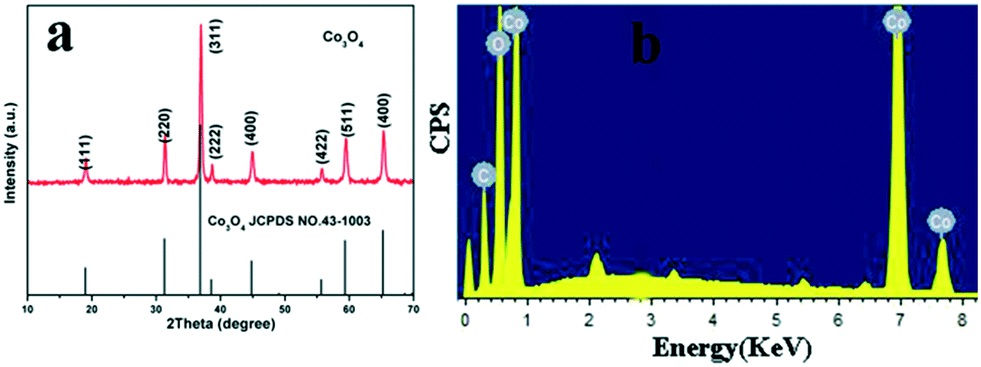 | ||
| Fig. 2 XRD pattern (a) and EDS spectrum (b) of Co3O4 microspheres after calcination at 500 °C. | ||
3.2. Effect of the amount of citric acid, PEG-400 and DMF on the morphology and microstructure of metal oxide products
In order to investigate the effects of the amount of citric acid, PEG-400 and DMF on the morphology and microstructure of the metal oxide products, the Co3O4 system was used as a representative and some control experiments were carried out. The morphology and microstructure of the Co3O4 samples with different molar ratios of citric acid to Co2+ were compared. The SEM and TEM images of the synthesized products are shown in Fig. 3. Without the addition of citric acid, the Co3O4 product displays irregular and block particles. When the molar ratio of citric acid to Co2+ is 3:4, the main product is a monolith with macropores (Fig. 3b). When the molar ratio of citric acid to Co2+ is 5:4, Co3O4 hollow core–shell microspheres can be obtained (Fig. 3c). Upon further increasing the molar ratio of citric acid to Co2+, the product is still hollow core–shell microspheres (not shown), whose morphology and microstructure exhibit no obvious change. However, too much citric acid will result in no precipitation. It is obvious that the appropriate amount of citric acid is responsible for the formation of Co3O4 microspheres.
 | ||
| Fig. 3 SEM images of the as-prepared Co3O4 samples obtained with different molar ratios of citric acid to Co2+ after calcination at 500 °C: (a) 0:4, (b) 3:4 and (c) 5:4. | ||
In order to examine the function of PEG-400 in the fabrication of metal oxide architectures, the SEM images of the Co3O4 samples prepared with different volumes of PEG-400 after calcination at 500 °C are shown in Fig. 4. Upon increasing the volume of PEG-400 from 0 ml to 10 ml while maintaining the other parameters, the Co3O4 samples all present well-dispersed microspheres (Fig. 4a and b), but the diameter of the microspheres obviously increases. On the other hand, the TEM images show that the Co3O4 microspheres prepared in the absence of PEG-400 possess a solid structure while the microspheres prepared with 10 ml of PEG-400 have a hollow core–shell structure (insets in Fig. 4a and b). Further increasing the volume of PEG-400 will lead to a large amount of precipitate in a short time, which results in the formation of irregular Co3O4 particles (Fig. 4c). Therefore, it is clear that the addition of PEG-400 is responsible for the formation of the hollow structure of the Co3O4 samples.
 | ||
| Fig. 4 SEM and TEM images of the Co3O4 samples obtained with various volumes of PEG-400 after calcination at 500 °C: (a) 0 ml, (b) 10 mL and (c) 15 mL. | ||
To investigate the effect of DMF on the morphology and microstructure of the product, the SEM and TEM images of the Co3O4 samples prepared with different volumes of DMF are shown in Fig. 5. It can be seen that the Co3O4 sample in the absence of DMF is mainly composed of a large number of irregular blocks. When 5 ml of DMF is added, Co3O4 hollow core–shell microspheres with diameters in the range of 1.0–5.0 μm can be observed (Fig. 5b). Further increasing the volume of DMF to 15 mL does not cause a significant change in the morphology of the product, and hollow core–shell structures are still the main product (Fig. 5c). However, the hollow core–shell structures become more uniform and their average diameter shows an increasing trend along with the increase of the volume of DMF. Therefore, it is clear that the presence of DMF is a necessary condition for the formation of monodispersed microspheres.
 | ||
| Fig. 5 SEM images of the as-prepared Co3O4 samples obtained with various volumes of DMF after calcination at 500 °C: (a) 0 mL, (b) 5 mL and (c) 15 mL. | ||
3.3. Formation mechanism of hollow core–shell structures
In order to investigate the state of coordination in the compound, the IR spectra of different Co3O4 precursor samples are given in Fig. 6a. The IR spectra of the three Co3O4 precursor samples show bands around 1588 cm−1 and 1412 cm−1, which are attributed to asymmetric (νas(COO)) and symmetric (νs(COO)) vibrations of coordinated carboxyls, respectively. Obviously, the frequency difference between νas(COO) and νs(COO) is 176 cm−1, indicating the bridge-bonding of carboxylate to two cobalt atoms.24,25 This revealed that the Co(II) cations can bind to citric acid, assembling into a polynuclear complex. The two new weak bands at 2960 cm−1 and 2865 cm−1 are found in the IR spectrum of the Co3O4 precursor sample synthesized without PEG-400, which are attributed to the asymmetric and symmetric vibrations of CH3.26 Thus, it can be considered that DMF exists in the precursor sample and combines with the formed polynuclear complex in the process of precipitation. On the other hand, there is an obvious absorption peak at 1132 cm−1 attributed to the stretching vibration of C–O–C of PEG-400 in the IR spectrum of the Co3O4 precursor sample synthesized without DMF, which proves that the molecular chain of –(OCH2CH2)n– in the PEG-400 molecules also combines with the formed polynuclear complex in the process of precipitation.27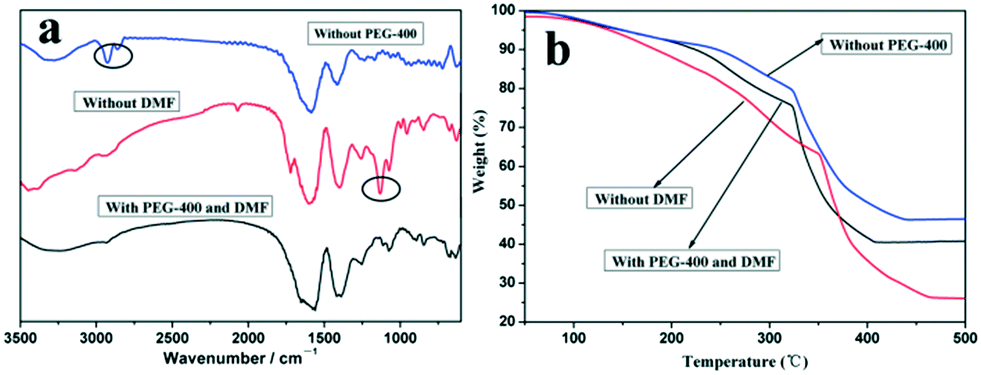 | ||
| Fig. 6 IR spectra (a) and TG curves (b) of different Co3O4 precursor samples. | ||
In order to investigate the effects of the presence of PEG-400 and DMF on the thermal properties of the Co3O4 precursors, the TG curves of three different Co3O4 precursors are given in Fig. 6b. There are obvious differences among the three TG curves. The sample prepared with only DMF has a smaller weight loss during the calcination process while that prepared with only PEG-400 has a larger weight loss compared to that prepared with DMF and PEG-400. On the other hand, the weight loss of the sample prepared with DMF and PEG-400 stopped at the lowest temperature among the three TG curves. It is clear that the presence of PEG-400 and DMF in the precursors causes synergistic effects on the thermal properties of the Co3O4 precursor sample, thus further affecting the morphology and microstructure of the Co3O4 products.
In order to monitor the formation process of the hollow core–shell structure, the samples prepared at different calcination temperatures were characterized by SEM, TEM and XRD. The SEM and TEM images (Fig. S3a and b†) show that the Co3O4 precursor samples without calcination are solid microspheres and have imperforated surfaces. There are no obvious pores between nanoparticles composing the precursor microspheres. It is obvious that the porous structure of the Co3O4 microspheres is formed during the heat treatment process as a result of the decomposition of the organics. The sample prepared at 300 °C still presents solid structures (Fig. S3c†). However, hollow core–shell microspheres (Fig. S3d†) are formed when the sample is prepared at 400 °C. The XRD patterns of the Co3O4 samples prepared at different calcination temperatures are given to track their phase composition and structure and shown in Fig. S4.† When the sample is prepared at 200 °C, no obvious diffraction peaks are observed in the XRD pattern, which confirms that the product exhibits an amorphous phase. When the sample is prepared at 300 °C, it is found that all the diffraction peaks can be readily indexed to those of the cubic spinel phase of Co3O4 except for a diffraction peak at 42.4° belonging to the (200) reflection of cubic CoO. Upon further increasing the calcination temperature to 400 °C, the diffraction peak of CoO still exists. When the temperature is increased to 500 °C, no other peaks from possible impurities are detected and the pure Co3O4 product is observed. In addition, the crystallization of the as-obtained Co3O4 samples is enhanced gradually along with the increase of heat treatment temperature.
On the basis of the above results, a plausible formation mechanism is schematically illustrated in Scheme 1. In solution, citric acid firstly coordinates to Co(II), assembling into a citrate–Co(II) complex. According to a previous report, the citrate–Co(II) complex in aqueous solution will eventually form a transparent sol rather than undergo precipitation along with the evaporation of solvent when there are no PEG-400 and DMF in the system.28 Therefore, the addition of PEG-400 and DMF into the solution facilitates the precipitation of the citrate–Co(II) complex at room temperature, which brings about the original nucleation and primary growth of amorphous nanoparticles. As a nonionic surfactant, PEG-400 molecules contain hydrophilic –O– moieties and hydrophobic –CH2–CH2– in the polymeric chain. When PEG-400 is added into the solution, there are interactions between the PEG-400 molecules, metallic ions and citrate, which make long chains of PEG-400 inter-twist with each other, with Co2+ ions in the middle. This coordination attraction among the PEG-400 molecules, metallic ions and citrate has been proved to facilitate the assembly of citrate–Co2+–PEG-400 molecules into complex nanoparticles.29,30 If DMF is not added, the very fast nucleation rate does not provide the formed nuclei with enough time for self-assembly, which leads to the formed nuclei randomly combining to form an irregular particle (shown in Fig. 4c). When DMF is added into the solution, its function is quite different from its conventional function in the DMF/H2O solvent, and it does not act as a mere solvent. The presence of DMF reduces the reactivity and mobility of the Co2+ ions, resulting in a sluggish crystallization process, which is considered favourable for synthesizing the kinetically stable phase.31,32 DMF can be adsorbed on the surface of citrate–Co2+–PEG-400 to facilitate the assembly of nanoparticles into complex nanostructures.33,34
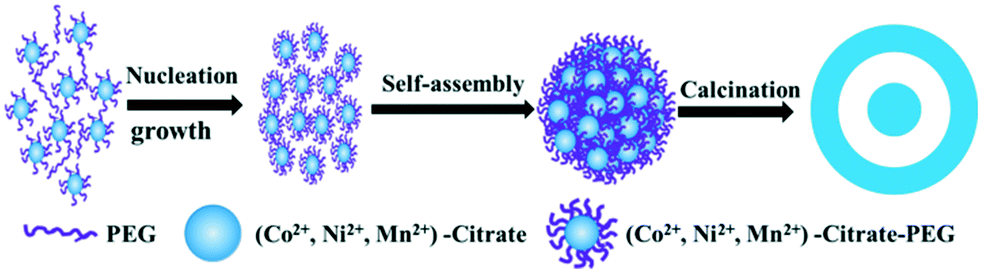 | ||
| Scheme 1 Schematic illustration of the evolution process of the obtained metal oxide microspheres. | ||
In the following sintering process, the high calcination temperature brings about a temperature gradient along the radial direction of the Co3O4 precursor microspheres, which causes the outermost layer to firstly change into a stiff outer shell. After the formation of the stiff outer shell, the Co3O4 microspheres will have the ability to resist a certain intensity of contraction. The inner core as the gel phase detaches from the stiff outer shell, which creates a void space between the outer shell and the inner core due to the temperature gradient along the radial direction. Along with the decomposition of a large amount of organic matter, the different contraction rates between the outer shell and inner core will further lead to the separation of the outer shell and the inner core, which finally results in the formation of hollow structures during the calcination process. When the temperature gradient is negligible, the heterogeneous contraction process by non-equilibrium heat transfer is terminated.35–37 During the calcination process, the release of a large amount of gas from the shell causes the shell to be porous rather than dense (insert in Fig. 1a). On the other hand, the combustion of PEG-400 occluded in the precursor also plays a key role in the formation of the hollow core–shell structure, which provides the Co3O4 microspheres with enough space to restructure during the hollow structure formation process. Therefore, it can be found that the Co3O4 microspheres prepared in the absence of PEG-400 possess solid structures while those prepared with PEG-400 display hollow core–shell structures (inserts in Fig. 4).
3.4. Electrochemical performances of metal oxide hollow core–shell microspheres
The applications of the as-synthesized metal oxide microspheres in energy conversion and storage were demonstrated. As anode materials for Li-ion batteries, the as-synthesized Co3O4 hollow core–shell microspheres were investigated as a typical example. Their electrochemical performance was evaluated by cyclic voltammetry (CV) and galvanostatic charge–discharge cycling. The cyclic voltammograms collected at a scan rate of 0.5 mV−1 between 0.01 and 3.0 V are shown in Fig. 7a. In the first cathodic scan, the sharp current peak at about 1.05 V is attributed to the reduction process of Co3O4 to metallic cobalt, the formation of amorphous Li2O and the formation of SEI layers.38,39 This reduction reaction has a significant effect on the reversible capacity of Co3O4.40,41 For the anodic scan, the broad peak at around 2.08 V is associated with the oxidation reactions and conversion of metallic cobalt into cobalt oxide. In the second cycle, the peak intensity of the main reduction peak apparently decreases and the oxidation peak becomes wider with weaker intensity. The CV curves are quite similar after the first cycle, indicating the good reversibility of the electrochemical reactions, and this is further confirmed by the following cycling performance test. Fig. 7b gives the corresponding galvanostatic discharge–charge curves within a voltage window of 0.01–3.0 V after the 1st, 2nd, 5th and 10th cycles. The first discharge voltage profile is for a typical Co3O4 electrode and there are two obvious voltage plateaus at about 1.25 and 1.10 V. The initial plateau is due to the formation of rock salt CoO and amorphous Li2O while the latter is related to the transition of the intermediate into metallic cobalt.42,43 In the charge process, the charge plateau located at about 2.0 V can be ascribed to the reversible oxidation of metallic Co into Co3O4, accompanied by the release of Li+.41 Afterwards, the following charge–discharge curves are very similar, implying that the electrochemical reactions proceed with stable cycling stages. In the first cycle, an initial discharge capacity of 1470 mA h g−1 and a charge capacity of 1025 mA h g−1 are observed at the current density of 100 mA g−1 with a coulombic efficiency of 69.7%. The irreversible capacity is attributed to the isolation and cracking of the Co3O4 hollow core–shell microspheres during the lithiation and delithiation processes, the irreversible formation of SEI layers in the discharge process and the incomplete decomposition of Li2O during the first charge.41,44 In the 5th cycle, the electrode shows a discharge capacity of 1200 mA h g−1 and a charge capacity of 1200 mA h g−1 with a coulombic efficiency of 100%. To further verify the performances of the hollow core–shell Co3O4 electrode, Fig. 8a shows the discharge capacity retention at a higher current density of 0.2 A g−1 after 80 cycles. After the first cycle, there is a marked increase in the specific capacities with the maximum value of 1125 mA h g−1 during the 50th cycle. Then, the specific capacity is retained at about 1100 mA h g−1 during the 80th cycle. Actually, the gradual increase of specific capacity at the beginning of the charge–discharge cycles is a common phenomenon for Co3O4-based anode materials, and the increasing capacity of the Co3O4 electrode in the first 50 cycles may be related to interfacial reactions like decomposition of the electrolyte and the formation of a gel-like layer, which promotes the activation and stabilization of the electrode.45,46 In contrast, the discharge capacity retention of solid Co3O4 microspheres obtained without PEG-400 after calcination at 500 °C was investigated and the corresponding discharge capacity retention at a current density of 0.2 A g−1 after 80 cycles is shown in Fig. 8a. The solid Co3O4 electrode suffers from rapid capacity fading and the discharge capacity decreases from 1126 mA h g−1 to 498 mA h g−1 after 80 cycles. It was clearly found that the Co3O4 hollow core–shell microspheres showed much better lithium storage capacity and cycling performance than the solid sample.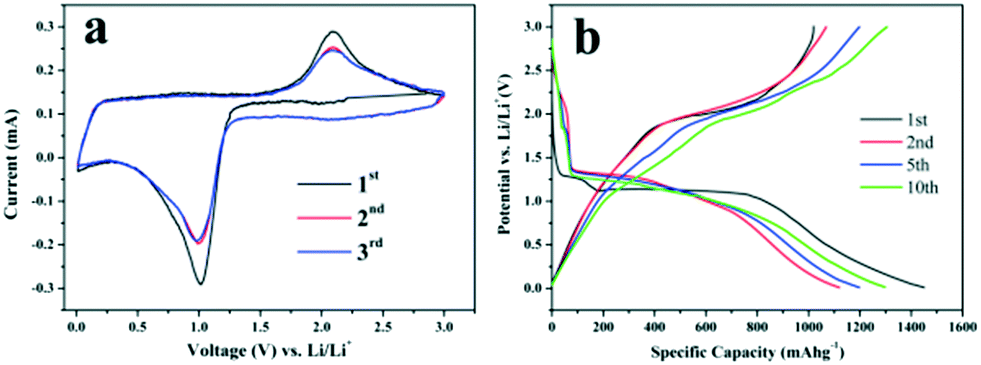 | ||
| Fig. 7 Cyclic voltammetry curves of Co3O4 hollow core–shell microspheres at a scan rate of 0.5 mV s−1 for the first three cycles (a) and their discharge–charge voltage profiles after the 1st, 2nd, 5th and 10th cycles at a current density of 0.1 A g−1 (b). | ||
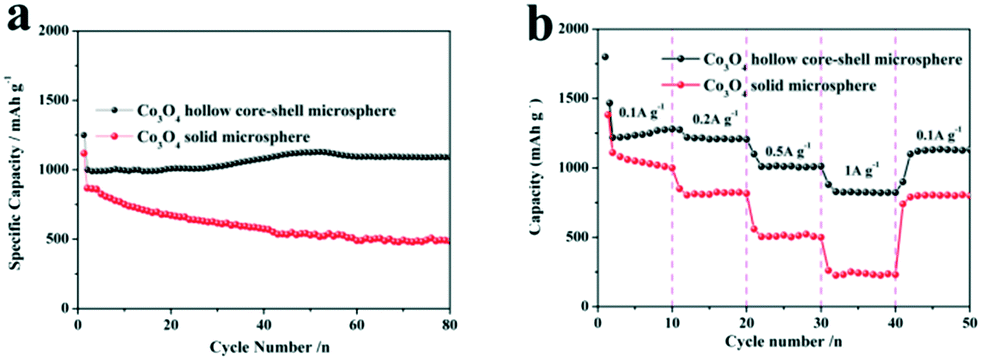 | ||
| Fig. 8 The discharge cycling performance versus cycle number of Co3O4 hollow core–shell microspheres and solid microspheres at a current density of 0.2 A g−1 (a) and the rate capabilities of the above two anodes at higher current varied from 0.1 A g−1 to 1.0 A g−1. | ||
Good high-C-rate performances are desirable for developing high power/fast charging lithium ion batteries. The batteries were cycled at various current densities of 0.1 A g−1, 0.2 A g−1, 0.5 A g−1 and 1.0 A g−1 (Fig. 8b). The Co3O4 hollow core–shell microspheres show high 2nd-cycle discharge capacities of 1220 mA h g−1, 1241 mA h g−1, 1103 mA h g−1 and 875 mA h g−1 at various current densities of 0.1 A g−1, 0.2 A g−1, 0.5 A g−1 and 1.0 A g−1, respectively. More importantly, a high 2nd-cycle capacity of 1105 mA h g−1 can be recovered rapidly when the current rate is reduced again from 1.0 A g−1 to 0.1 A g−1. In contrast, the specific capacities of the Co3O4 solid microspheres drop rapidly at increased current densities (Fig. 8b). For example, the Co3O4 solid microspheres show a discharge capacity of only about 256 mA h g−1 at 1.0 A g−1. Therefore, it is clear that the hollow core–shell microspheres simultaneously exhibit high specific capacity and stability compared to the solid microspheres, which is ascribed to their special structure. The hollow structure can increase the number of active reaction sites and the volumetric capacity of the metal oxide microspheres. On the other hand, the hollow core–shell structure provides an elastic buffer space to effectively accommodate the volume expansion that occurs and prevents aggregation and the electrical isolation of metal oxides during the cycling process. Importantly, the thin shells and the nano-sized units facilitate the rapid electrochemical reaction kinetics by enhancing lithium diffusion and electron conduction to accommodate fast cycling rates.21,47,48
3.5. Adsorption properties using Congo red
Due to the universality and toxic nature of dye pollutants, the removal and separation of dye pollutants has become very important for wastewater treatment. In this paper, as an example of potential applications, the as-prepared hierarchical NiO microspheres were used as an adsorbent for wastewater treatment, and experiments on the adsorption performance towards Congo red were carried out. A certain amount of the NiO product is dispersed into Congo red solution with an initial concentration of 60 mg L−1 without any other additives. The UV-vis absorption spectra of Congo red in the presence of the NiO hollow core–shell microspheres after different time intervals are given in Fig. 9a and show the characteristic intense peak at around 500 nm in aqueous solution. The absorption peak intensity clearly decreases upon using the NiO hollow core–shell microspheres as adsorbents and equilibrium can be established within 120 min, which indicate that the adsorption of Congo red on the NiO hollow core–shell microspheres is efficient. The relative adsorption capacity of NiO was evaluated by C/C0 (C0 and C represent the concentrations of Congo red before and after treatment, respectively) and the corresponding curve is given in Fig. 9b. It can be seen that the NiO hollow core–shell microspheres can remove ca. 75% of Congo red for an initial concentration of 60 mg L−1 solution after 180 min contact time.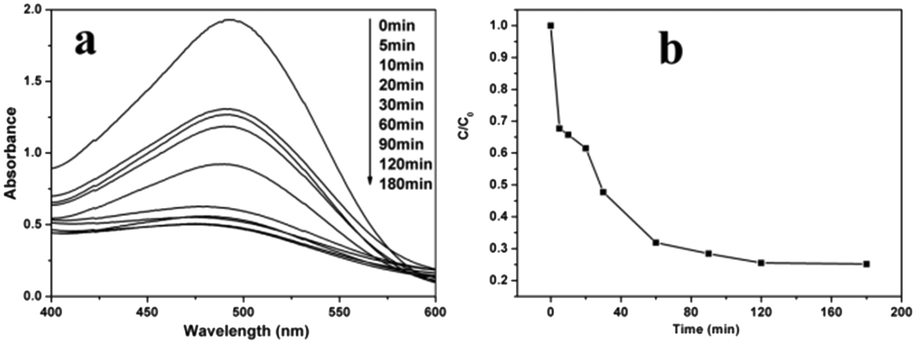 | ||
| Fig. 9 UV-vis absorption spectra of Congo red in the presence of NiO hollow core–shell microspheres after different time intervals (a) and the adsorption rate of Congo red on the NiO hollow core–shell microspheres (b); the initial concentration of Congo red is 60 mg L−1. | ||
The equilibrium isotherm of a specific adsorbent represents its adsorptive characteristics and is very important to the design of adsorption processes. The common models used to investigate the adsorption isotherm are the Langmuir and Freundlich equations, which are expressed as follows:49,50
Langmuir model: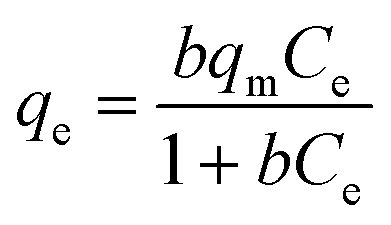
Freundlich model: qe = KfCe1/n where the constant b is the Langmuir affinity constant (L mg−1), qm is the maximum adsorption capacity (mg g−1), Kf is roughly an indicator of the adsorption capacity, and 1/n is the Freundlich exponent (dimensionless). The experimental results of this study were fitted with these two models. Experiments for the estimation of the adsorption isotherms of Congo red on the NiO microspheres were performed. The adsorption isotherms of Congo red on the sample were examined with an adsorption time of 4 h, and the corresponding Langmuir and Freundlich plots are given in Fig. 10. The model parameters are listed in Table 1. The high squares of the correlation coefficients of 0.9964 and 0.9804, respectively, reveal that the adsorption of the samples toward Congo red fits the Langmuir model better. For the NiO hollow core–shell microspheres, the maximum adsorption capacity is as high as 95.28 mg g−1. Solid NiO microspheres synthesized without PEG are prepared and their corresponding SEM and TEM images are given in Fig. S5a.† As a comparison, their adsorption performance was also evaluated using Congo red as the adsorbate. Fig. S5b† shows the UV-vis absorption spectra of Congo red in the presence of solid NiO microspheres after different time intervals. The intensity of the absorption peaks clearly decreases upon using NiO solid microspheres as the adsorbent and equilibrium can be established within 120 min. The adsorption rate of Congo red on the solid NiO microspheres is also shown in Fig. S6a.† It can be seen that the absorption peak intensities decrease when the NiO solid microspheres are used as adsorbents, indicating that they can remove ca. 50% of Congo red at an initial concentration of 60 mg L−1 solution after 180 min contact time. The corresponding Langmuir and Freundlich plots are given in Fig. S6b and the model parameters are listed in Table S1.† A maximum adsorption capacity of 63.73 mg g−1 can be obtained, which is lower than that of the hierarchical NiO hollow core–shell microspheres. It is considered that the intrinsic hollow core–shell nature as well as the high porosity of the shell contributes greatly to the improvement in their performance as adsorbents for the removal of Congo red from wastewater.
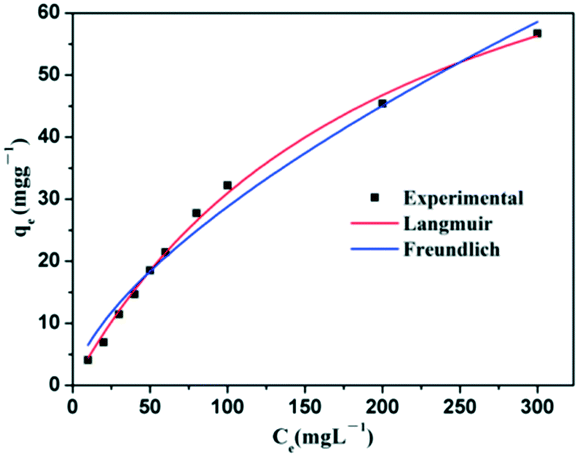 | ||
| Fig. 10 Adsorption isotherm curves for the adsorption of Congo red onto NiO hollow core–shell microspheres. | ||
| Isotherm models | Constants | Value |
|---|---|---|
| Langmuir | b (Lm g−1) | 0.0048 |
| q m (mg g−1) | 95.2829 | |
| R 2 | 0.9964 | |
| Freundlich | K f | 1.4664 |
| n | 1.5467 | |
| R 2 | 0.9804 |
4. Conclusions
In summary, an ultra-facile coordination precipitation route followed by a calcination process has been developed to synthesize Co3O4, NiO and Mn2O3 hollow core–shell microspheres for the first time. The amount of citric acid, PEG and DMF (or glycerol) has important effects on the formation of metal oxide hollow core–shell microspheres, and their possible formation mechanisms are proposed based on experimental parameters. The formation from the precursor to the hollow core–shell microspheres involves the original nucleation and primary growth of nanoparticles, and their self-assembly, as well as mass transportation from the interior to the exterior of the microspheres. Calcination plays an important role in controlling the hollow structure formation, and the combustion of PEG provides the metal oxide nanoparticles with ample space for restructuring. As anode materials for lithium ion batteries, the as-synthesized Co3O4 hollow core–shell microspheres exhibited enhanced electrochemical performances compared to solid microspheres. Furthermore, the as-obtained NiO product exhibited good adsorption properties for Congo red during water treatment and the corresponding maximum adsorption capacity was 95.28 mg g−1 at room temperature. It is expected that this ultra-facile approach can be extended to the synthesis of other polynary metal oxides for energy conversion, storage applications and adsorption.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 51102138, 51372129, 51372125 and 51572138).Notes and references
- J. Li, J. Wang, X. Liang, Z. Zhang, H. Liu, Y. Qian and S. Xiong, ACS Appl. Mater. Interfaces, 2014, 6, 24–30 CAS
.
- X. Li, L. Wang, J. Shi, N. Du and G. He, ACS Appl. Mater. Interfaces, 2016, 8, 7276–17283 Search PubMed
- L. Dang, H. Ma, J. Xu, Y. Jin, J. Wang, Q. Lu and F. Gao, CrystEngComm, 2016, 18, 544–549 RSC
- J. Du, J. Qi, D. Wang and Z. Tang, Energy Environ. Sci., 2012, 5, 6914–6918 CAS
- H. Wu, S. Zhang, J. Zhang, G. Liu, J. Shi, L. Zhang, X. Cui, M. Ruan, Q. He and W. Bu, Adv. Funct. Mater., 2011, 21, 1850–1862 CrossRef CAS
- J. Qi, K. Zhao, G. Li, Y. Gao, H. Zhao, R. Yu and Z. Tang, Nanoscale, 2014, 6, 4072–4077 RSC
- J. Lee, S. H. Hwang, J. Yun and J. Jang, ACS Appl. Mater. Interfaces, 2014, 6, 15420–15426 CAS
- J. Zhang, Y. Cao, C. Wang and R. Ran, ACS Appl. Mater. Interfaces, 2016, 8, 8670–8677 CAS
- B. Lu, A. Liu, H. Wu, Q. Shen, T. Zhao and J. Wang, Langmuir, 2016, 32, 3085–3094 CrossRef CAS PubMed
- L. Peng, H. Zhang, L. Fang, Y. Bai and Y. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 4745–4753 CAS
- S. Zhang, F. Ren, W. Wu, J. Zhou, X. Xiao, L. Sun, Y. Liu and C. Jiang, Phys. Chem. Chem. Phys., 2013, 15, 8228–8236 RSC
- A. M. El-Toni, M. A. Habila, J. P. Labis, Z. A. Alothman, M. Alhoshan, A. A. Elzatahry and F. Zhang, Nanoscale, 2016, 8, 2510–2531 RSC
- X. Wang, Y. Bai, Q. Zhang and Y. Yin, Chem. Rev., 2016, 116, 10983–11060 CrossRef CAS PubMed
- X. Lai, J. E. Halperta and D. Wang, Energy Environ. Sci., 2012, 5, 5604–5618 CAS
- G. Zhang and X. W. David Lou, Angew. Chem., Int. Ed., 2014, 53, 9041–9044 CrossRef CAS PubMed
- X. Lai, J. Li, B. A. Korgel, Z. H. Dong, Z. Li, F. Su, J. Du and D. Wang, Angew. Chem., Int. Ed., 2011, 50, 2738–2741 CrossRef CAS PubMed
- L. Zhou, H. Xu, H. Zhang, J. Yang, S. B. Hartono, K. Qian, J. Zou and C. Yu, Chem. Commun., 2013, 49, 8695–8697 RSC
- Y. J. Xiong, B. Wiley, J. Chen, Z. Li, Y. Yin and Y. Xia, Angew. Chem., Int. Ed., 2005, 44, 7913–7917 CrossRef CAS PubMed
- X. Wang, X. Wu, Y. Guo, Y. Zhong, X. Cao, Y. Ma and J. Yao, Adv. Funct.
Mater., 2010, 20, 1680–1686 CrossRef CAS
- K. Y. Niu, J. Park, H. Z. M. Zheng and A. P. Alivisatos, Nano Lett., 2013, 13, 5715–5719 CrossRef CAS PubMed
- S. Xu, C. M. Hessel, H. Ren, R. Yu, Q. Jin, M. Yang, H. Zhao and D. Wang, Energy Environ. Sci., 2014, 7, 632–637 CAS
- L. Han, R. Liu, C. Li, H. Li, C. Li, G. Zhang and J. Yao, J. Mater. Chem., 2012, 22, 17079–17085 RSC
- F. Qu, H. Jiang and M. Yang, Nanoscale, 2016, 8, 16349–16356 RSC
- J. E. Crowell, J. G. Chen and J. T. Yates Jr, J. Electron Spectrosc. Relat. Phenom., 1986, 39, 97–106 CrossRef CAS
- R. J. Jouet, A. D. Warren, D. M. Rosenberg, V. J. Bellitto, K. Park and M. R. Zachariah, Chem. Mater., 2005, 17, 2987–2996 CrossRef CAS
- Z. G. Hu, P. Prunici, P. Patzner and P. Hess, J. Phys. Chem. B, 2006, 110, 14824–14831 CrossRef CAS PubMed
- H. Liang, Y. Pei, J. Li, W. Xiong, Y. He, S. Liu, Y. Lia and B. Li, RSC Adv., 2016, 6, 31374–31385 RSC
- Y. Gu, F. Jian and X. Wang, Thin Solid Films, 2008, 517, 652–655 CrossRef CAS
- X. Chen, X. Jing, J. Wang, J. Liu, D. Song and L. Liu, CrystEngComm, 2013, 15, 7243–7249 RSC
- H. Zeng, W. Cai, P. Liu, X. Xu, H. Zhou, C. Klingshirn and H. Kalt, ACS Nano, 2008, 2, 1661–1670 CrossRef CAS PubMed
- J. Zhang, B. Yao, H. Ping, Z. Fu, Y. Li, W. Wang, H. Wang, Y. Wang, J. Zhang and F. Zhang, RSC Adv., 2016, 6, 472–480 RSC
- X. Guo, S. Yu and G. Cai, Angew. Chem., Int. Ed., 2006, 45, 3977–3981 CrossRef CAS PubMed
- J. Xu and Y. Zhu, CrystEngComm, 2012, 14, 2702–2710 RSC
- J. Xu and Y. Zhu, CrystEngComm, 2011, 13, 5162–5169 RSC
- S. Yu, Q. Yao, G. Zhou and S. Fu, ACS Appl. Mater. Interfaces, 2014, 6, 10556–10565 CAS
- J. Qi, X. Lai, J. Wang, H. Tang, H. Ren, Y. Yang, Q. Jin, L. Zhang, R. Yu, G. Ma, Z. Su, H. Zhao and D. Wang, Chem. Soc. Rev., 2015, 44, 6749–6773 RSC
- H. Shin, W.-J. Lee and J. Mater, Chem. A, 2016, 4, 12263–12272 CAS
- S. Qiu, H. Gu, G. Lu, J. Liu, X. Li, Y. Fu, X. Yan, C. Hua and Z. Guo, RSC Adv., 2015, 5, 46509–46516 RSC
- L. Li, K. H. Seng, Z. Chen, Z. Guo and H. Liu, Nanoscale, 2013, 5, 1922–1928 RSC
- K. J. Lee, T.-H. Kim, T. K. Kim, J. H. Lee, H.-K. Song and H. R. Moon, J. Mater. Chem. A, 2014, 2, 14393–14400 CAS
- B. Wang, X. Lu and Y. Tang, J. Mater. Chem. A, 2015, 3, 9689–9699 CAS
- D. Larcher, G. Sudant, J. Leriche, Y. Chabre and J. Tarascon, J. Electrochem. Soc., 2002, 149, A234–A241 CrossRef CAS
- C. Hu, J. Guo, J. Wen and Y. Peng, J. Mater. Sci. Technol., 2013, 29, 215–220 CAS
- F. Zheng, K. Shi, S. Xu, X. Liang, Y. Chen and Y. Zhang, RSC Adv., 2016, 6, 9640–9646 RSC
- Z. Zhang, L. Li, Q. Xua and B. Cao, RSC Adv., 2015, 5, 61631–61638 RSC
- X. Rui, H. Tan, D. Sim, W. Liu, C. Xu, H. H. Hng, R. Yazami, T. M. Lim and Q. Yan, J. Power Sources, 2013, 222, 97–102 CrossRef CAS
- J. Wang, N. Yang, H. Tang, Z. Dong, Q. Jin, M. Yang, D. Kisailus, H. Zhao, Z. Tang and D. Wang, Angew. Chem., 2013, 125, 6545–6548 CrossRef
- H. Lin, H. Rong, W. Huang, Y. Liao, L. Xing, M. Xu, X. Li and W. Li, J. Mater. Chem. A, 2014, 2, 14189–14194 CAS
- L. Ai and Y. Zeng, Chem. Eng. J., 2013, 215/216, 269–278 CrossRef
- B. Cheng, Y. Le, W. Cai and J. Yu, J. Hazard. Mater., 2011, 185, 889–897 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ce02563c |
| This journal is © The Royal Society of Chemistry 2017 |