
Overview of the strategic approaches for the solid-state recognition of hydrated anions
Md. Najbul
Hoque†
and
Gopal
Das
 *
*
Department of Chemistry, Indian Institute of Technology Guwahati, Assam 781 039, India. E-mail: gdas@iitg.ernet.in
First published on 8th February 2017
Abstract
The recognition of hydrated anions is a rapidly expanding area in the domain of supramolecular chemistry. Since the beginning of the research field of the supramolecular chemistry of anions, ample literature studies on the recognition of anions have become available. However, there is less literature on the solid-state recognition of hydrated anions in comparison. This highlight covers recent advances in the solid-state recognition of hydrated anions. Structural variation of the anion–water cluster is primarily governed by the shape and charge density of the anions. Given the interest in anion–water interactions, various structural assemblies of anion–water clusters are discussed in this highlight. The review includes both purely organic and metal–organic systems that are potentially suitable for stabilizing anion–water clusters. The recognition of hydrated anions by artificial ligands has attracted huge research interest for potential application in waste remediation, catalysis and transport. Due to their indigenous nature of a high propensity of hydration, inorganic anions can be crystallized with various degrees of hydration; however, their structures are very poorly understood. This review thus gathers the articles published concerning the recently developed field of hydrated anions. This short review might be helpful for researchers to gain an insight into some design features of ligands and anion–water interactions.
 Md. Najbul Hoque | Dr Najbul Hoque was born in West Bengal, India. He received his BSc (2008) and MSc (2010) in Chemistry from Krishnath College, University of Kalyani and the Indian Institute of Technology Delhi, respectively. He earned his PhD in Supramolecular Chemistry under the supervision of Prof. Gopal Das in 2015 from the Indian Institute of Technology Guwahati. Currently, he is working as a postdoctoral fellow with Prof. Ming-Liang Tong at Sun Yat-Sen University, P. R. China. His current research focuses on the preparation of guest-dependent Hofmann-type spin-crossover materials for potential applications in molecular switches, chemical/physical sensors and memory devices. |
 Gopal Das | Prof. Gopal Das received his MSc degree in 1993 from Calcutta University, India. He obtained his PhD in 1999 from the Indian Institute of Technology Kanpur under the supervision of Prof. Parimal K. Bharadwaj. Subsequently, he moved to the University of Geneva, Switzerland as a postdoctoral research associate in the group of Prof. Stefan Matile. In 2004, he joined the Indian Institute of Technology Guwahati as an Assistant Professor and in 2013 he became a Full Professor. He has published more than 150 research papers to date and his current research interests are in the field of supramolecular chemistry and crystal engineering. |
1. Introduction
The design and synthesis of ligands for the recognition of inorganic anions is of considerable interest due to its immense application potential in the domain of biomedical and environmental sciences.1–6 It is now well established that for suitable anion coordination, the presence of a H-bonding donor site, such as an amide, amine, urea or thiourea, or sometimes pyrrole, indole or guanidinium, as a recognition element is critical in receptors.7–10 A major challenge in anion recognition by synthetic ligands is the need to organize appropriate binding sites for a particular anionic guest selectively from its highly hydrated environment. As an example, the knowledge acquired from the current research trend in this field has been successfully implemented in the separation of sulfate anions from water with nuclear waste.11,12 In general, most inorganic anions remain partially hydrated during crystallization with the host molecule owing to their high solvation energies. Their extreme water affinity is facilitated by multiple hydrogen bonds. Despite the tremendous progress in anion coordination chemistry over the past decade, there are only a few publications in the literature involving solid-state structural studies that describe various anion–water interactions of hydrated anions in crystalline solids. The complex hydration nature of anions in an aqueous medium due to the unique properties of water, such as polarity, H-bonding abilities, etc., make the structures of anion–water clusters very unpredictable.13,14 It has also been established that the reactivity of a hydrated ion is quite different from that of bare anions or anions in nonpolar media. A molecular level understanding of hydrated anions is required through elucidation of the solute–solvent interactions. Theoretical studies on anion–water assemblies primarily suggested that anion–water clusters are predominantly controlled by H-bonding and other weak noncovalent supramolecular interactions arising from the surrounding ligand.15–21 The structural characterization and analysis of the energetics of anion–water cluster formation are essential for understanding the solvation processes, ion translocation in water–membrane interfaces, ion mobilities in the bulk, and the electrical phenomena that occur within aqueous–salt interfaces.22,23 One could gain a clear insight of both water–water and anion–water interactions through the solid-state structural elucidation of anion–water clusters. The design of a ligand with complementary H-bonding sites will guide us to synthesize new ligands for anions. Though less progress has been made on the solid-state recognition of solvated anions, except for some serendipitous discovery of anion–water clusters in predesigned molecular systems such as in the effective sulfate separation from seawater through the crystallization of sulfate–water clusters,24,25 it has been also found in the literature that hydrated anions play a vital role in stabilizing the formation of capsular assemblies by filling the larger void present in the system. We do believe that there is much more to be done to carry forward these new concepts and to delve into applications to real-world problems. Since the advances made in the supramolecular chemistry of anions, a number of reviews on synthetic ligands covering various anionic species have appeared in recent years.26–38In recent years, we have witnessed a number of reports of hydrated anion clusters appearing in the literature, but no efforts have been made yet towards the collection of those in a review format. This endeavour is the first attempt to make a comprehensive record on the unique structural correlation among various inorganic anions and water molecules. The aim of this highlight article is to obtain an overview on hydrated anion recognition within organic and metal-complex-based systems with a special emphasis on inorganic anions. In addition, careful attention is also paid to the interesting design tricks employed to ensure the stability of these clusters, with more emphasis given to the report published in the last few years. Efforts have been made to offer a comprehensive overview of anion–water clusters, including spherical, planar, and tetrahedral inorganic anions and a few organic ions. Since the final goal in this area of research is to elucidate the anion–water structure, we limited our survey to systems dealing with pure anion binding. We have divided our report on the basis of the shape of anions. The general colour code followed in this manuscript is as follows: C, purple; N, blue; O, red; F, light green; Cl, deep green; Br, brown; I, violet; S, yellow; P, light brown; and H, magenta.
2. Anion–water assemblies of spherical anions
The most challenging targets for the recognition of spherical halide anions lies in their high hydration enthalpy.38 The dynamic and disordered nature of halogen anions in various aqueous environments make it difficult to understand the exact degree of hydration as well as the structure and reactivity of these anions. For example, in case of fluoride ions, owing to their small size and high hydration enthalpy, recognition of the hydrated fluoride ion is targeted preferably rather than the naked ion.38 In addition, water fluoridation in drinking water causes dental and skeletal fluorosis, which demands detailed study of the hydrated fluoride anion.2.1 Hydrated fluoride ion
One successful approach to designing a ligand for an anion is to synthesize molecules that offer an array of H-bonding sites, i.e. ligands with polyamine, amide, urea or thiourea functionality. Water molecules, present in the crystal system, sometimes act as a secondary H-bonding motif towards anions, and interestingly generate unique anion–water clusters. The elucidation and prediction of structures belonging to this category still remains a daunting task. In most of the cases, anions are partially hydrated, with the degree of hydration varying from ligand to ligand.Ghosh and coworkers described ligands for the recognition of the hydrated F− ion or other anions. Most of the ligands formed a bowl-shaped conformation with a dimeric assembly and a wide space inside the cavity, which facilitates binding with hydrated anions rather than bare anions. A benzene-capped tripodal amide ligand L1 with a pyridyl moiety exhibited hydrated anion recognition in aqueous media.39L1 in the presence of tetrabutylammonium fluoride (TBAF) yielded a hydrated F− complex containing a tweezer-like parallelogram-shaped fluoride–water cluster [F2–(H2O)4]2−. The cluster was embedded into the dimeric assembly of ligand L1 (Fig. 1b), which was further propagated to form a 1D-polymer thorough a pyridyl moiety. The average H-bonding distance in the cluster was found to be 2.798 Å. The cluster was stabilized mainly by two N–H⋯F, six O–H⋯F and four N–H⋯O interactions.
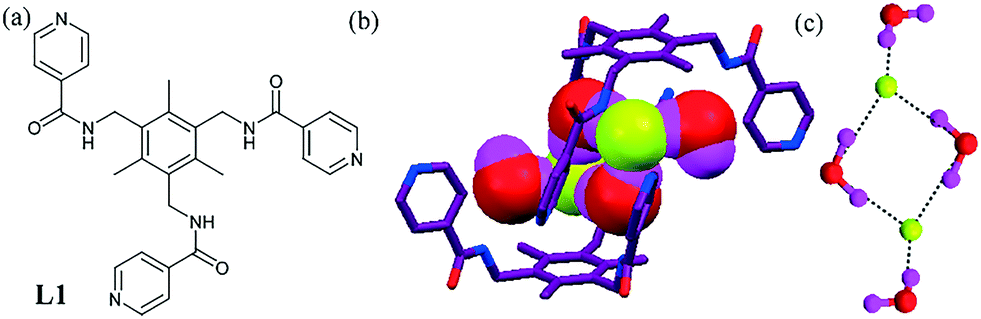 | ||
| Fig. 1 (a) Molecular structure of L1. (b) Encapsulation of the [F2–(H2O)4]2− cluster in L1. (c) Fluoride–water cluster [F2–(H2O)4]2− obtained on treatment of L1 with TBAF. Reproduced from ref. 39, with permission from the Royal Society of Chemistry (CCDC reference number 933028). | ||
The same group also reported ligands (L2 and L3) functionalized with a nitrophenyl group, which could recognize anions in various hydrated states. The o-NO2-substituted ligand L2 formed a bowl-shaped cavity, and then two such bowls could intercalate to form a dimeric capsule (Fig. 2b and d).40 The addition of TBAF yielded a dimeric capsule with an encapsulated hydrated F− ion. The H-bonding pattern in the cluster [F2–(H2O)6]2− showed that it consisted of three tricyclic arrangements, where two F− ions were in the apex of each cycle, bridged by two water dimers and two water molecules (Fig. 2c).
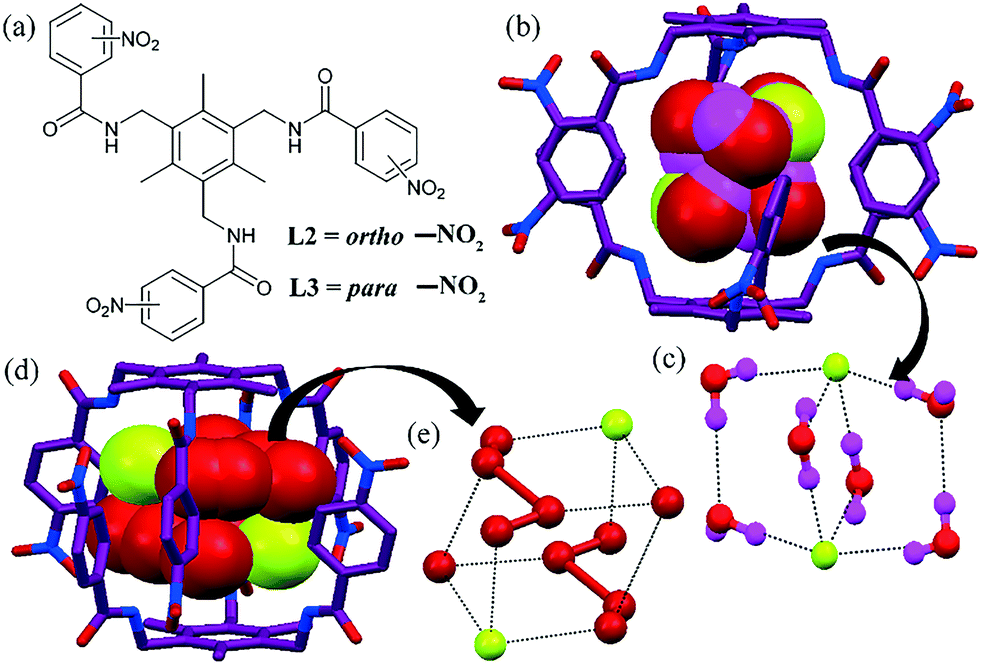 | ||
| Fig. 2 (a) Molecular structures of L2 and L3. (b) The [F2–(H2O)6]2− cluster in L2. (c) Close view of the [F2–(H2O)6]2− cluster in L2. (d) The [F2–(H2O)6]2− cluster in L3. (e) Close view of the [F2–(H2O)6]2− cluster in L2. Fig. 2(a–c) are reproduced from ref. 40, with permission from the Royal Society of Chemistry. Fig. 2(d and e) is reprinted (adapted) with permission from ref. 41. Copyright 2010 American Chemical Society (CCDC reference number 733118 and 732741). | ||
The chloride and nitrate complex of L2 failed to yield this anion–water assembly or dimeric complex formation. Anion-dependent aggregation experiments with F−, NO3− and AcO− proved the selective F−-assisted formation of capsular assembly over other planar anions. However, p-nitro-substituted L3 rendered the encapsulation of two NO3−, [(AcO)2–(H2O)4]2−, [F2–(H2O)6]2−, and [Cl2–(H2O)4]2−, respectively, in a dimeric capsular assembly,41 where the fluoride–water cluster was isostructural (stoichiometrically) to the previous case with little difference in the arrangement of F− and water (Fig. 2e).
Inspired by tripodal ligands, an intelligent synthesis by the same group yielded a hexapodal ligand on a benzene platform, which formed a big cage for the encapsulation of larger anion–water clusters. An o-CF3 substituted hexaamide42 ligand L4 formed a chair-like conformation, and then an amazing conformational change occurred during hydrated F− anion recognition by creating a large cavity (Fig. 3b). The [F4–(H2O)6]4− cluster was recognized by the dimeric cage of L4 (Fig. 3b and c). Herein, two [F2–(H2O)2]2− clusters were bridged by two water molecules within the dimeric capsular assembly, which stabilized the thermodynamically disfavored aaaaaa conformation. In the case of the AcO− complex, the ligand retained the aaabbb conformation of free L5. In the NO3− complex, L4 adopted an irregular aabaab conformation and it was recognized in a tetrapodal cleft. In the AcO− complex, a similar aaabbb conformation was observed, wherein each AcO− anion was encapsulated in two independent tripodal clefts without any water of crystallization.
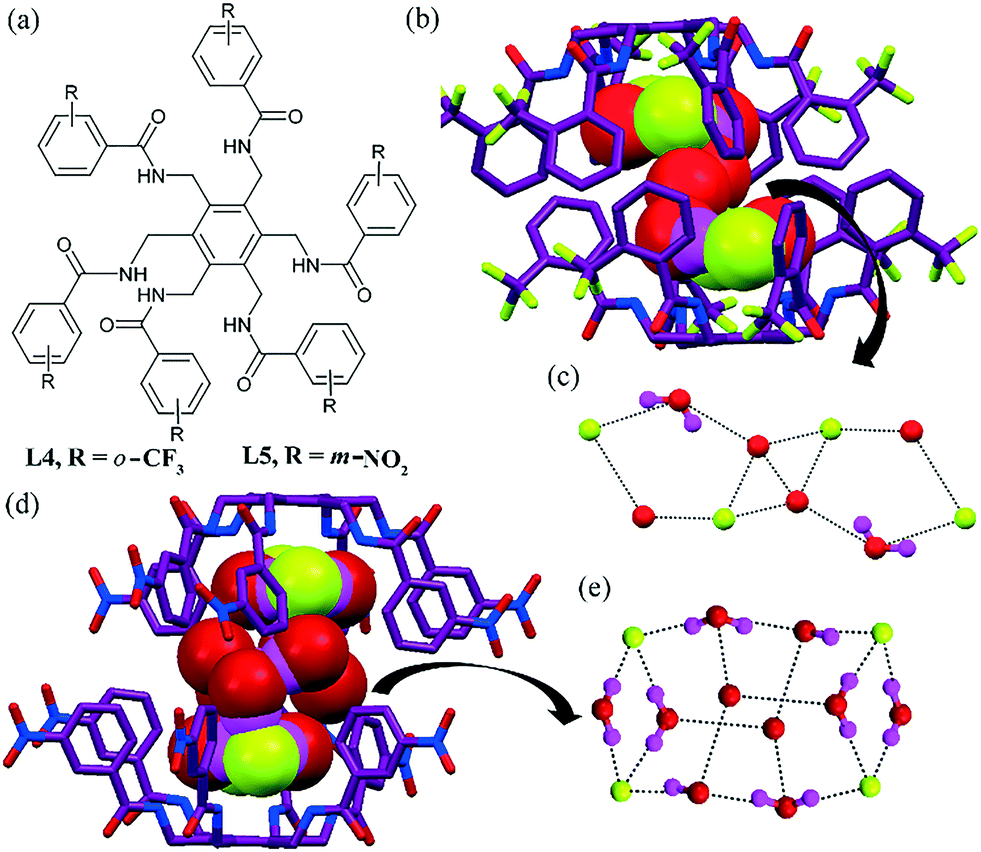 | ||
| Fig. 3 (a) Molecular structures of L4 and L5. (b) Encapsulation of the fluoride–water cluster [F4–(H2O)6]4− within the capsular assembly of L4. (c) Close view of the [F4–(H2O)6]4− cluster. (d) Space-filled view of the [F4–(H2O)10]4− cluster inside the capsular assembly of L5. (e) Close view of the [F4–(H2O)10]4− cluster. Reproduced from ref. 42 and 43, with permission from the Royal Society of Chemistry (CCDC reference numbers 996875 and 812289). | ||
The solid-state structure of the fluoride complex of the ligand L5 revealed the encapsulation of an unusual fluoride–water cluster, [F4–(H2O)10]4−, inside the dimer with the highly unfavorable aaaaaa conformation of the hexapodal bowl (Fig. 3d and e).43 In this case, all the arms of L5 were projected uni-directionally to engulf the fluoride–water cluster inside the dimeric assembly of the ligand. Fig. 3e depicts the H-bonding pattern of the [F4–(H2O)10]4− cluster formed via strong fluoride–water interactions. The cluster contains four-, five- and six-membered rings, constructed by H-bonding interactions. The cyclic hexameric water cluster showed a chair conformation and O⋯O distances ranging from 2.737 Å to 2.902 Å (average: 2.823 Å). The cluster contained various O–H⋯F interactions with average O⋯F bond distances of 2.683 Å ranging from 2.628 Å to 2.728 Å. Thus, the encapsulated [F4–(H2O)10]4− cluster was constructed and stabilized by various H-bonding interactions such as N–H⋯F, N–H⋯O, O–H⋯F, O–H⋯O, C–H⋯F and C–H⋯O interactions. In the AcO− complex, the aaabbb conformation was stabilized and each AcO− anion was encapsulated in two independent tripodal clefts without any water of crystallization.
Bowman-James and coworkers reported another big tricyclic amide-based ligand as a versatile host, particularly for hydrated anions.44 The tetrahedron L6 could capture a tetrahedrally arranged fluoride–water cluster [F–(H2O)4]− (Fig. 4a–c). The ligand also encapsulated a water pentamer similar to a fluoride–water cluster, where the central water formed four hydrogen bonds (two acceptors and two donors). Though the geometry of the two clusters was the same, as the F− ion was a four H-bond acceptor, the orientation of the two water molecules was inverted. The O⋯F distance ranged from 2.585 to 2.615 Å. Therefore, this dynamic switching of a two H-bond donor and acceptor (water pentamer) to a four H-bond acceptor (fluoride–water cluster) within a microenvironment is very important in biological systems where bond making and breaking are very common. Solution-state NMR anion binding studies have also shown preferable F− binding by the ligand L6, and this is helpful in predicting the solvation of anions in such an isolated environment.
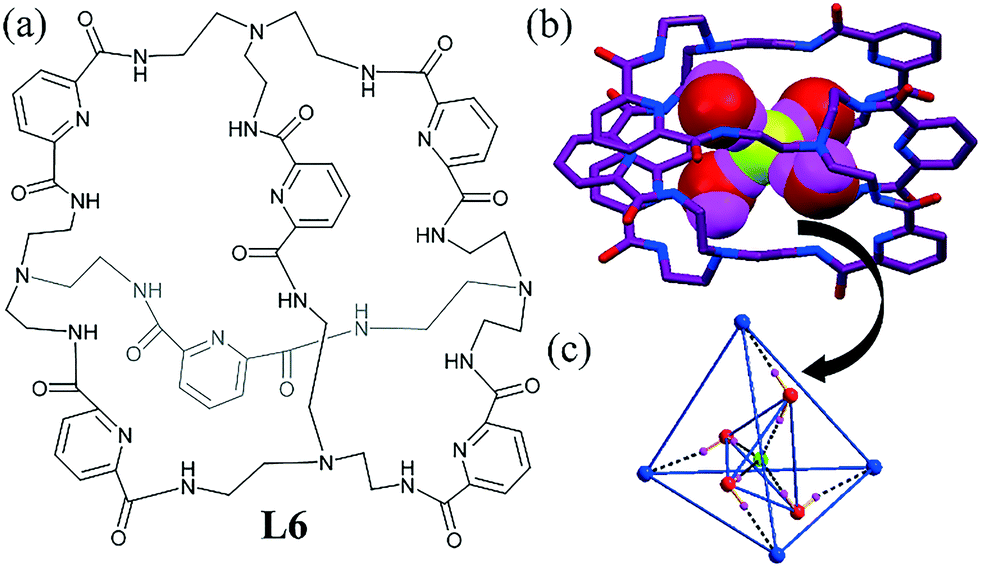 | ||
| Fig. 4 (a) Molecular structure of L6. (b) Tetrahedral fluoride–water cluster [F–(H2O)4]− within the tetrahedral cage L6. (c) Tetrahedral cluster within a tetrahedral core. Reproduced with permission from ref. 44. Copyright Wiley-VCH Verlag GmbH & Co. KGaA (CCDC reference number 838464). | ||
The cleft-shaped ligand L7 decorated with amine and amide functionality could selectively detect F− ions in solution and recognize hydrated fluoride ions in the solid state (Fig. 5b).45 The crystal structure of the fluoride complex revealed that between the two F− ions, one was pseudo-encapsulated within the cleft, whereas the other interacted with water to form a fluoride–water cluster [F2–(H2O)3]2−. The F− ion in the cleft remained a little outside of the mean molecular plane of L7 with a distorted square planar geometry, while the other F− behaved in an entirely different manner to form a H-bonded fluoride–water cluster, which mediated the formation of the ligand dimer. The cluster was stabilized by various N–H⋯F and N–H⋯O H-bonding interactions (1.992–2.910 Å).
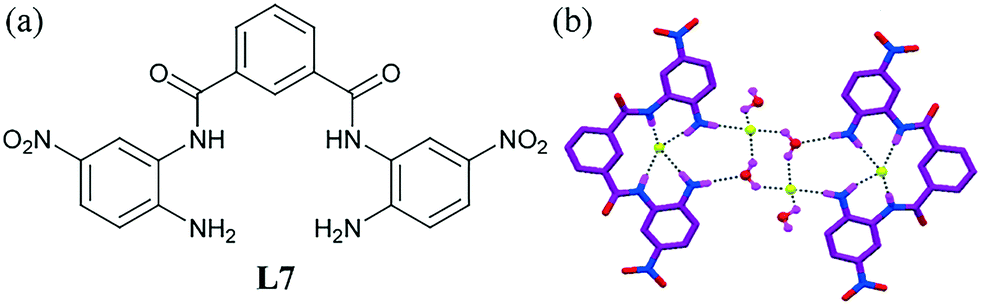 | ||
| Fig. 5 (a) Molecular structure of L7. (b) View of the fluoride–water cluster [F2–(H2O)3]2− and encapsulation of the fluoride ion by the cleft-shaped ligand L7. Non-bonding hydrogen atoms and tetrabutylammonium (TBA) counter cations are omitted for clarity. Reproduced from ref. 45, with permission from the Royal Society of Chemistry (CCDC reference number 842781). | ||
Recent advances in anion recognition show that a tripodal ligand can produce ample H-bonding sites and can demonstrate very interesting anion–ligand interaction. A N4-platform-based polyamine tripodal ligand L8 reported by our group possessed multiple protonation sites (Fig. 6a).46L8 produced stable complexes with almost all inorganic anions, ranging from spherical to tetrahedral. All the three arms were pointed in the same direction and formed a C3v symmetric cavity. All three amine groups and the apical N-atom were protonated in the presence of an acid, which consequently produced a highly ionic environment leading to the formation of a capsular/non-capsular hydrated anion cluster. For instance, the fluoride complex of L8 formed a long chain acyclic fluoride–water cluster, [F7–(H2O)8]7−. Herein, two open chains consisting of four water molecules and three fluoride ions are symmetrically distributed on both sides of the central F1 ion leading to the formation of the cluster (Fig. 6b). It was observed that the [F7–(H2O)8]7− cluster played an important role in aggregation of the cationic ligand [H4L8]4+ in a capsular fashion (Fig. 6c). The hybrid anion–water cluster was established to act as a molecular belt in stitching the two cationic tripodal ligands together.
 | ||
| Fig. 6 (a) Molecular structure of L8. (b) Acyclic fluoride–water chain [F7–(H2O)8]7−. (c) Fluoride–water belt-induced bimolecular capsule. Reproduced from ref. 46, with permission from the Royal Society of Chemistry (CCDC reference number 961079). | ||
The recognition of hydrated F− ions has been accomplished even by cyclic ligands, which provide a microenvironment to trap guest species. Though the ligand L9 could encapsulate the hydrated F− anion inside its cavity, it formed a cuboid molecular box consisting of the protonated ligand, SIF62− and water molecules and containing a fluoride–water cluster inside the molecular box (Fig. 7b).47 The F− complex was assembled in a highly ordered fluoride–water cyclic octamer, [F–(H2O)]44− between two protonated L9 ligands (Fig. 7c). The bonding patterns were surprisingly similar to that for the cyclic [H2O]8 cluster reported by Atwood et al.48 with four water molecules as donors and four water molecules as acceptors.
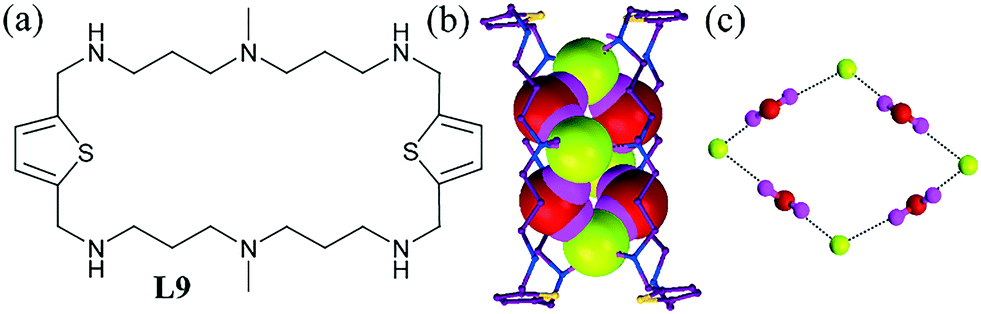 | ||
| Fig. 7 (a) Molecular structure of L9. (b) Space-filling view of the [F–(H2O)]44− cluster in a molecular box. (c) Close view of the fluoride–water cluster. Reprinted (adapted) with permission from ref. 47. Copyright 2012 American Chemical Society (CCDC reference number 860049). | ||
The cyclic azacryptand L9 could not encapsulate a hydrated F− ion, despite possessing a large cavity. However, there is another report of a p-xylyl-based azacryptand L10, which successfully captured two F− ions separated by a water molecule (Fig. 8b),49 where this was the first example of an “anion-based cascade complex”. In the “V” = shaped fluoride–water cluster, two F− ions reside almost in the center of the cavity (Fig. 8c). This finding helps to correlate between traditional transition metal coordination chemistry and anion coordination chemistry, where hydrogen bonding in the latter replaces lone-pair coordination in the former.
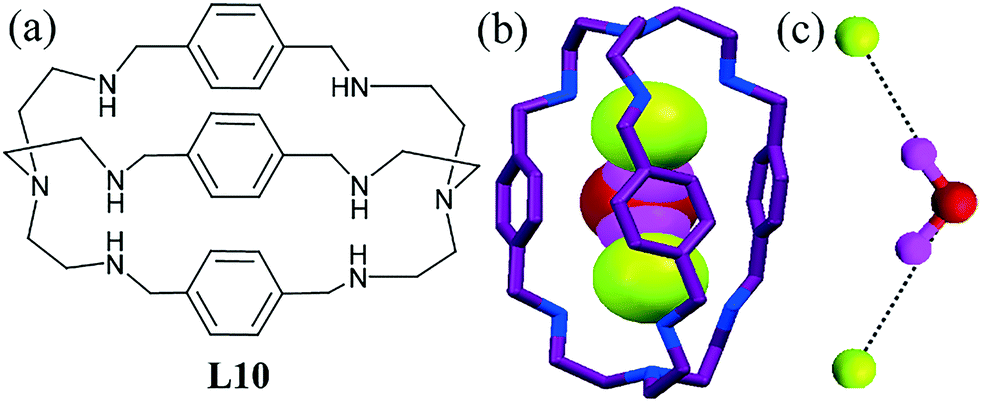 | ||
| Fig. 8 (a) Molecular structure of L10. (b) View of the water cascade in the cavity of L10. (c) V-shaped fluoride–water cluster [F–(H2O)2]−. Reproduced with permission from ref. 49. Copyright Wiley-VCH Verlag GmbH & Co KGaA (CCDC reference number 172118). | ||
More recently, our group reported two linear bis-urea ligands L11.50 Our investigation showed the entrapment of a fluoride–water cluster within the barrel-shaped assembly of the ligand (Fig. 9b). The cluster was made of two water molecules and two fluoride ions forming a cyclic fluoride–water tetramer [F2–(H2O)2]2− (Fig. 9c). The urea moiety and aromatic hydrogen were mainly coordinated with the anion–water cluster. Each fluoride ion in the cluster was coordinated by three urea molecules via strong N–H⋯F− and two water O–H⋯F− H-bonding interactions. The O⋯F bond was found to be very strong (2.5–2.8 Å), suggesting a very stable cyclic cluster tightly held in the crystal lattice of the ligand.
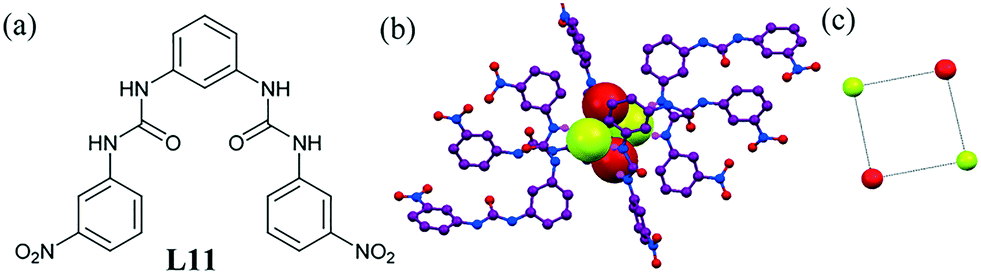 | ||
| Fig. 9 (a) Molecular structure of L11. (b) Entrapment of the fluoride–water cluster [F2–(H2O)2]2−. (c) Square-shaped fluoride–water cluster. Reprinted (adapted) with permission from ref. 50. Copyright 2016 American Chemical Society (CCDC reference number 1450226). | ||
2.2 Hydrated chloride anion
A conformationally flexible C3v symmetric acyclic ligand L12 containing H-bonding functionality was reported by our group.51 The extended ligand with a microcavity demonstrated the encapsulation of a discrete cyclic halide–water (chloride or bromide) tetrameric cluster. The tripodal ligand with a bridgehead nitrogen suitable for protonation produced an appropriate scaffold for anionic guests via H-bonding coordination. Two units of monohydrated chloride encapsulated L12 were held together by two Cl⋯H2O hydrogen bonds to form a dimeric capsular assembly (dN1⋯N1 = 20.621(3) Å) that encapsulated the [Cl2–(H2O)2]2− cluster in its center via H-bonding interactions. The H-bonding NH units of the ligand were widely spaced to offer an appropriate binding pocket for a single Cl− anion, resulting in the formation of an interesting [Cl2–(H2O)2]2− cluster encapsulated (Fig. 10b) within a dimeric capsular assembly.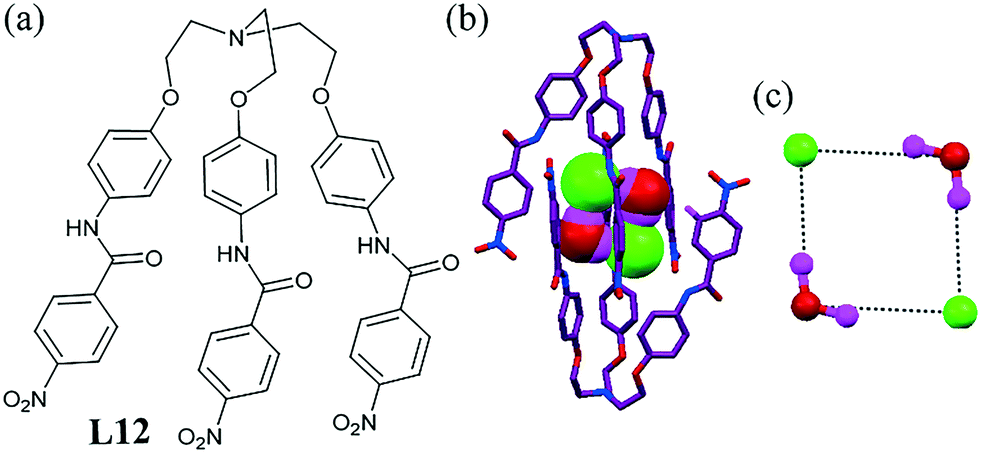 | ||
| Fig. 10 (a) Molecular structure of L12. (b) Encapsulation of the [Cl2–(H2O)2]2− cluster inside the dimeric capsular assembly. (c) Close up view of the chloride–water tetramer. Reproduced from ref. 51, with permission from the Royal Society of Chemistry (CCDC reference number 922581). | ||
The ligand L13 was designed to observe some steric interaction between isopropyl groups.52 Very surprisingly, discrete a dichloride–hexahydrate cluster was observed, in which the chloride anions occupied opposite corners of [Cl2–(H2O)6]− (Fig. 11c). Effectively, each chloride anion had another chloride ion in the third solvation sphere. Chloride ions were also involved in C–H⋯Cl− interactions with L13.
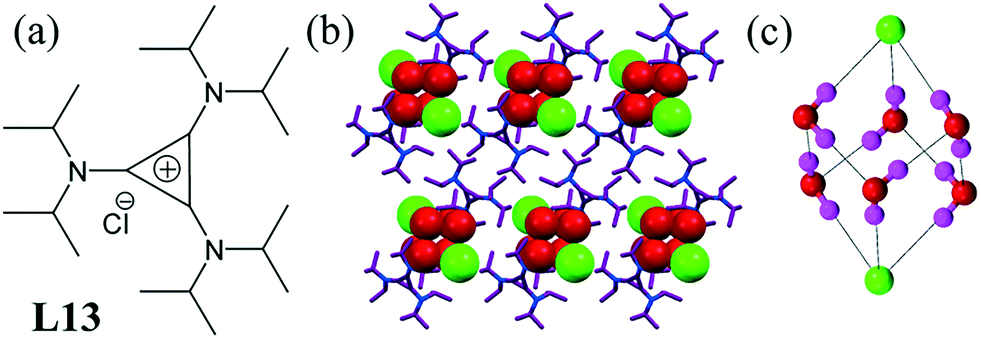 | ||
| Fig. 11 (a) Molecular structure of L13. (b) Chloride–water cluster [Cl2–(H2O)6]− surrounded by L13. (c) Cubane-like dichloride–hexahydrate cluster. Reproduced with permission from ref. 52. Copyright Wiley-VCH Verlag GmbH & Co. KGaA (CCDC reference number 615639). | ||
Another interesting assembly of chloride–water in an octaamino cryptand L10 (ligand structure described in Fig. 8a) was reported.53 The discrete propeller-shaped tricyclic water cluster [H2O]11 was also connected to an encapsulated Cl− ion (Fig. 12). The chloride–water cluster possessed a C3 axis passing through the encapsulated chloride ion, same as the ligand. The encapsulated chloride ion formed a strong H-bond with the apical N-atom and water molecules.
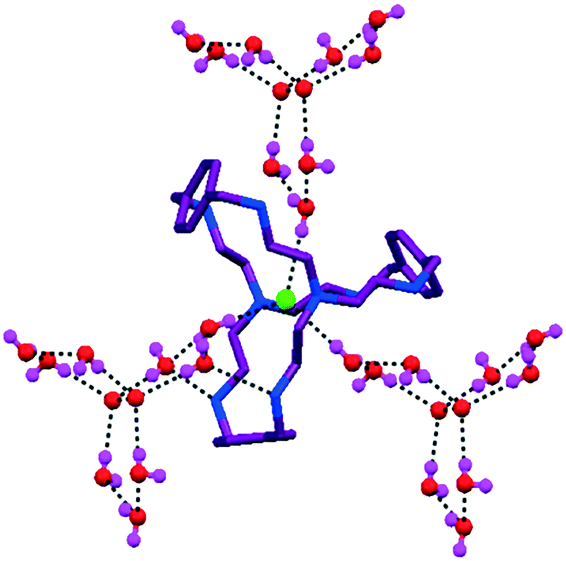 | ||
| Fig. 12 Propeller-shaped chloride–water cluster with an encapsulated Cl− ion in L10. Reproduced with permission from ref. 53. Copyright Wiley-VCH Verlag GmbH & Co. KGaA (CCDC reference number 295431). | ||
The chloride salt of L14 displayed various anion–water assemblies depending on the process of crystallization.54 It formed a 1D infinite zigzag water–chloride chain when crystallized from aqueous medium (Fig. 13b). However, crystallization in 50% acetic acid solution formed an unprecedented 2D water–chloride anionic net involving infinite [Cl6–(H2O)10]6− motifs (Fig. 13c). Herein, a cyclic centrosymmetric octameric structure [Cl2–(H2O)6]2− was the minimum ring size of the big cluster, while hydrogens of the terminal water oxygen acted as donors from the basic 16-membered centrosymmetric [Cl2–(H2O)6]2− motif. The O⋯Cl distances ranged from 2.32 to 2.45 Å and the average O⋯O distance was 2.863 Å. Thermal analysis also explained the stability of the hydrated anion cluster. The reversible solid-state dehydration and rehydration experiment showed a successful recovery of the [Cl6–(H2O)10]6− cluster in the hydrated organic cationic complexes. Moreover, a reversible inter-conversion of these two hydrated complexes also provided comprehensive information about the cooperative H-bonding behaviour.
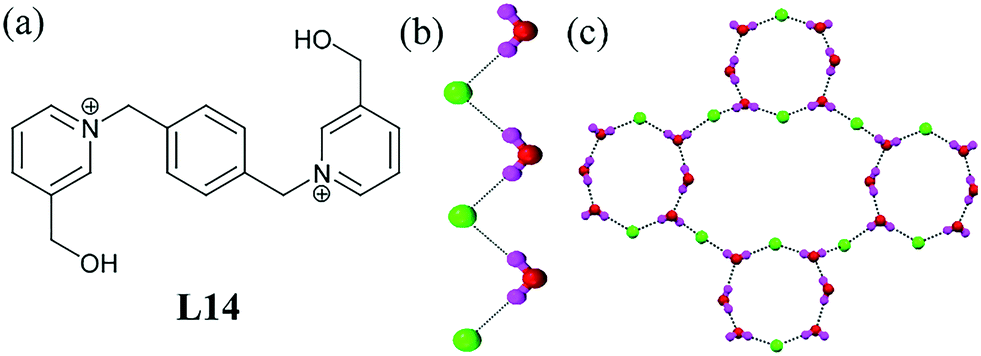 | ||
| Fig. 13 (a) Molecular structure of L14. (b) Zigzag water–chloride chains in the hydrated form of L14. (c) 2D infinite water–chloride network containing the [Cl2–(H2O)6]2− motif. Reprinted (adapted) with permission from ref. 54. Copyright 2012 American Chemical Society (CCDC reference numbers 838841 and 838842). | ||
A dichloro-substituted oxacalix[2]arene[2]triazine L15 led to the formation of an interesting chloride–water assembly stabilized by a few unconventional interactions, such as chloride⋯π interactions, along with lone-pair⋯π interaction.55 The ligand self-assembled into a rectangular cage and subsequently recognized a cyclic tetrameric chloride–water cluster [Cl2–(H2O)2]2− (Fig. 14b). The Cl− ion was located almost above the center of one of the triazine rings, while the distance of Cl− to the plane and the centroid of triazine was 3.238 and 3.258 Å, respectively. A water molecule formed a lone-pair⋯π interaction with the other triazine ring.
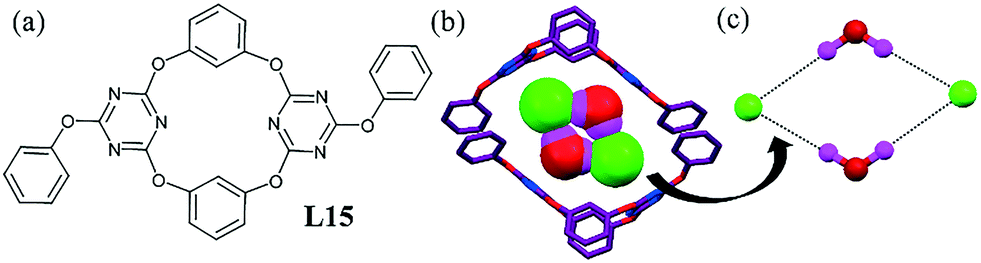 | ||
| Fig. 14 (a) Molecular structure of L15. (b) Inclusion of a chloride–water cluster [Cl2–(H2O)2]2− by ligand L15. (c) Close view of the H-bonded chloride–water tetramer. Reproduced from ref. 55, with permission from the Royal Society of Chemistry (CCDC reference number 897876). | ||
We reported a bowl-shaped tripodal ligand L16 that stabilized a hydrated chloride.56 The benzimidazole moiety acted as a protonation site and formed a complex with anions. The flexible cationic tripodal ligand provided the platform to host a chloride–water cluster [Cl3–(H2O)4]3− with a half-chair-like conformation (Fig. 15b). Each pentameric cluster, consisting of two chloride and three water molecules, was connected to the next pentamer unit by the tail, which had one chloride and one water molecule. The hydrated chloride–water lies between the hydrophobic channel of the ligand and the infinite chain passes thorough the hydrophobic channel in a zigzag fashion (Fig. 15c). The Cl⋯O contacts were in the range from 3.18 to 3.27 Å, which were consistent with the literature.
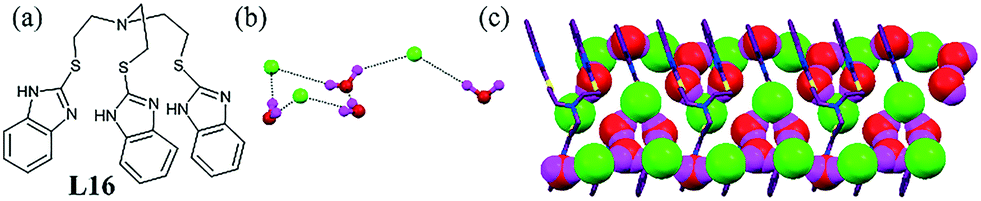 | ||
| Fig. 15 (a) Molecular structure of L16. (b) Chloride–water cluster [Cl3–(H2O)4]3− having a half-chair conformation. (c) Packing of a pentameric unit in a zigzag fashion. Reprinted (adapted) with permission from ref. 56. Copyright 2012 American Chemical Society (CCDC reference number 836926). | ||
Two isomeric metal complexes of the ligand L17 formed a 1D chloride–water chain with a chair-like architecture within the channel of these complexes.57 Three water molecules with a Cl− ion were involved in forming the infinite chloride–water cluster. The adjacent four-membered rings were composed of three water molecules and one Cl− ion (Fig. 16b). The O⋯O distances ranged from 2.583(3) to 2.852(3) Å in the chromium complex and 2.559(3)–2.855(3) Å in the iron complex in the anion–water cluster.
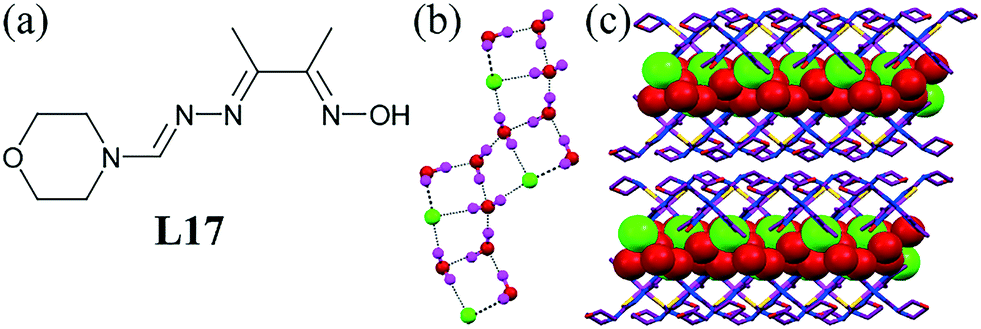 | ||
| Fig. 16 (a) Molecular structure of L17. (b) Chair-like topology of the chloride–water cluster. (c) Channels filled with chloride–water chains. Reproduced from ref. 57, with permission from the Royal Society of Chemistry (CCDC reference numbers 644970 and 644971). | ||
A MOF host was reported containing channels functionalized with free –COOH as H-bonding sites that could act as a selective host for a [Cl–(H2O)4]− cluster (Fig. 17c).58 The cationic MOF exhibited a perfect square-grid topology, with Cu2+ and L18 acting as nodes and linkers, respectively (Fig. 17b). Four water molecules were on the basal plane, while Cl− was placed on the vertex of the pyramidal chloride–water cluster. The Cl− was positioned on the 4-fold rotation axis, passing through the center of the channel with a Cl⋯O distance of 3.34 Å. The matching shape of the cluster and the cavity of MOF was the main driving force for the inclusion as competitive experiments with Cu(NO3)2, Cu(ClO4)2, or CuSO4 in place of CuCl2 strongly suggested an absence of the inclusion of other anions or an anion–water cluster. The inclusion of other halide ions was observed, as evident by IR spectra, due to the halide ions being of very similar size.
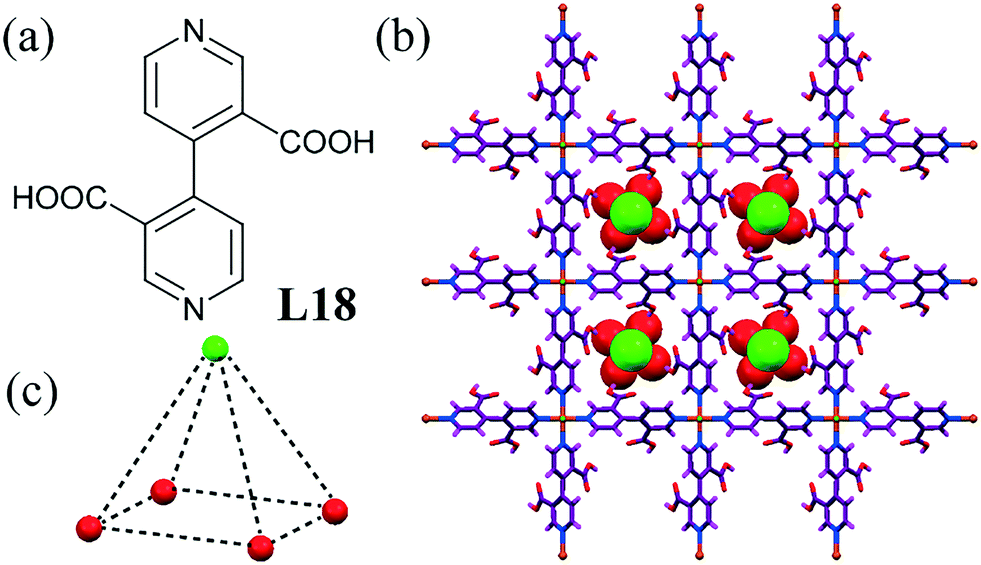 | ||
| Fig. 17 (a) Molecular structure of L18. (b) Square-grid network containing [Cl–(H2O)4]− clusters. (c) Square-pyramidal chloride–water cluster. Reprinted (adapted) with permission from ref. 58. Copyright 2005 American Chemical Society (CCDC reference number 261390). | ||
A metal–organic supramolecular host containing adipic acid and 1,10-phenanthroline (L19) could stabilize the formation of a [Cl2–(H2O)14]2− cluster.59 The cluster was formed by periodic repetition of a four-membered ring comprising two water molecules and two chloride ions. Another 12-membered ring was observed to contain 12 water molecules and two Cl− ions with a tetrameric water cluster (Fig. 18b). This anion–water cluster was arranged in the channel of the 3D supramolecular host.
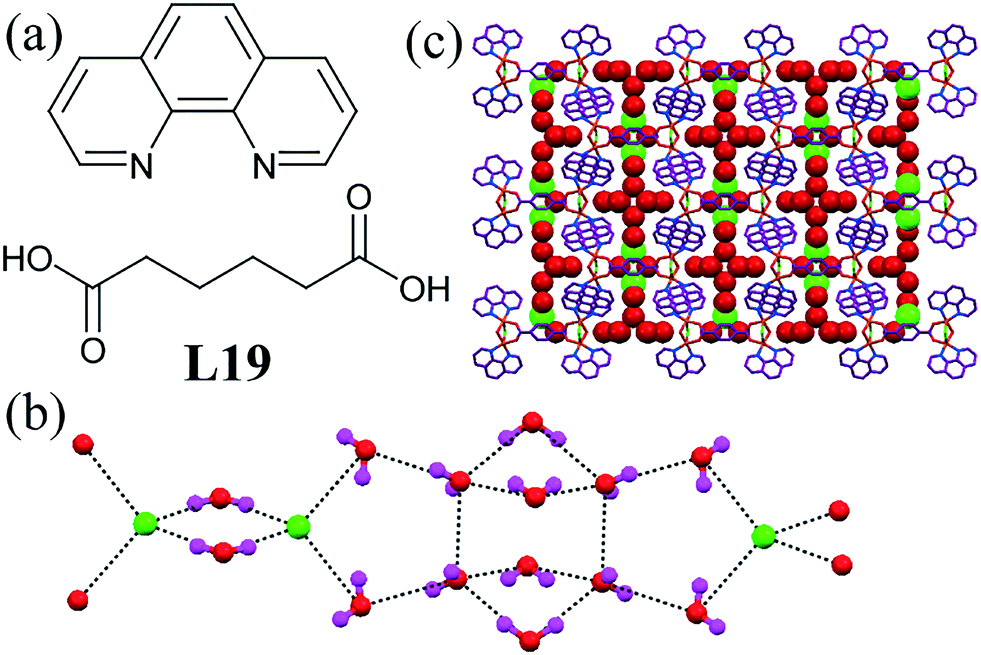 | ||
| Fig. 18 (a) Molecular structure of L19. (b) 1D assembly of [Cl2–(H2O)14]2−. (c) Chain of the chloride–water cluster. Reprinted (adapted) with permission from ref. 59. Copyright 2006 American Chemical Society (CCDC reference numbers 258089). | ||
The complexation of stannic salt and vitamin B6 (L20) could produce isostructural metal complexes with the incorporation of isostructural chloride/bromide–water clusters.60 The Cl− ions and the water molecules were involved in a network of H-bonds to create [Cl2–(H2O)4]2− tapes (Fig. 19b). In these tapes, planar tetrameric water clusters were linked by two chloride ions creating staircase-like architectures running parallel to the b axis. Alternatively, they could be described as a periodic repetition of fused 4-membered rings and 6-membered rings. In the hexameric cluster, each water molecule was H-bonded to two other water molecules and to a Cl− ion, whereas each Cl− was H-bonded to two water molecules.
 | ||
| Fig. 19 (a) Molecular structure of L20. (b) View of the [Cl2–(H2O)4]2− tape placed in the ab plane in the ligand L20. (c) Packing diagram showing the progress of the hexameric cluster through a crystal lattice. Reprinted from ref. 60, with permission from Elsevier (CCDC reference number 900734). | ||
Pombeiro et al. reported Ni(II) complexes of L21.61 The chloride salt of the Ni(II) complex formed a hexameric chloride–water cluster [Cl2–(H2O)4]2− (Fig. 20b). Two symmetry-generated chloride–water trimers were doubly interconnected to generate clusters composed of a cyclic planar tetranuclear core and two dangling water molecules. Within this cluster the O⋯O and O⋯Cl separations lay in the range of 2.706(9) Å to 3.172(7) Å.
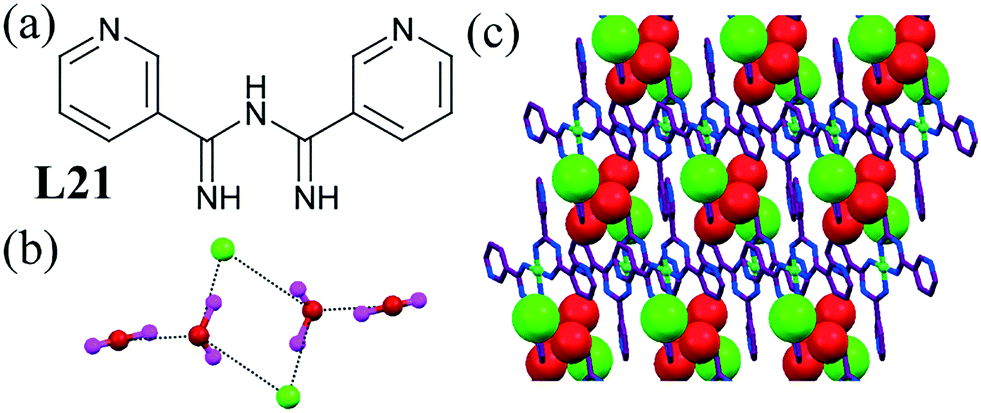 | ||
| Fig. 20 (a) Molecular structure of L21. (b) Packing diagram of [Cl2–(H2O)4]2− clusters (c) H-bonding pattern in chloride–water cluster. Reproduced with permission from ref. 61. Copyright Wiley-VCH Verlag GmbH & Co. KGaA (CCDC reference numbers 644970 and 644971). | ||
In another study, it was shown that a tetranuclear square-grid nickel complex made of the pyridine–urea-based ligand L22 could incorporate a chloride–water zigzag chain into its hydrophobic channel.62 The packing diagram of the square grids revealed both hydrophobic (aromatic rings) and hydrophilic channels (N–H atoms from the hydrazone groups) (Fig. 21b). Water and Cl− ion were situated in the hydrophobic channels, giving rise to 2D zigzag water–chloride chains, which were strongly held in the lattice through N–H⋯Cl H-bonds with two dangling –NH groups from the parallel ligand molecules.
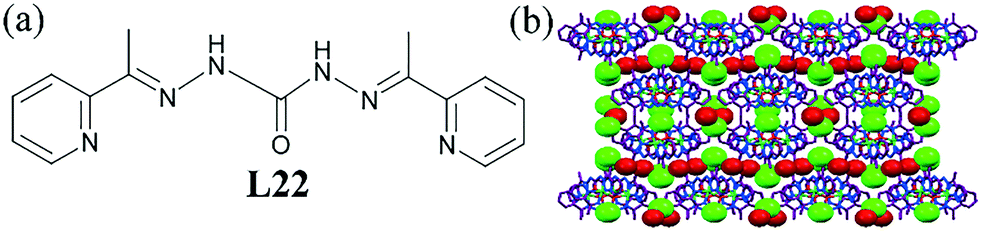 | ||
| Fig. 21 (a) Molecular structure of L22. (b) Packing diagram showing a hydrophobic channel filled up with chloride–water chains in ligand L22. Reprinted (adapted) with permission from ref. 62. Copyright 2013 American Chemical Society (CCDC reference number 941871). | ||
In a recent study, the isolation and structural characterization of a 2D water–chloride cyclic cluster [Cl8–(H2O)18]8− within a Ru-complex of L23 was reported.63 The crystal structure of complex L23 contained well-expanded H-bonding interactions between all the lattice water molecules and chloride anions, which basically led to the formation of the cluster. Each cyclic cluster was interconnected to the next cyclic part by the tail having a water molecule and Cl− ion (Fig. 22b). The cluster with repeating units contained various rings, such as hexamers and tetramers. All the cyclic rings were chloride–water hybrids with one or two chloride atoms.
 | ||
| Fig. 22 (a) Molecular structure of L23. (b) Cyclic cluster [Cl8–(H2O)18]8− in the Ru-complex of ligand L23. Reprinted from ref. 63, with permission from Springer (CCDC reference number 1430236). | ||
Pombeiro et al. contributed towards the understanding of the hydration phenomena of Cl− ions.64 They showed a unique 2D water–chloride anionic layer [Cl4–(H2O)20]4− within the crystal structure of the Fe(II) complex L24. Each of these clusters was further connected to grow a 2D layer structure (Fig. 23b). The 2D chloride–water layer occupied the free space between two hydrophobic arrays of the metal–organic unit with the help of a few C–H⋯O interaction. The stability of the hybrid cluster was also confirmed by thermal analysis.
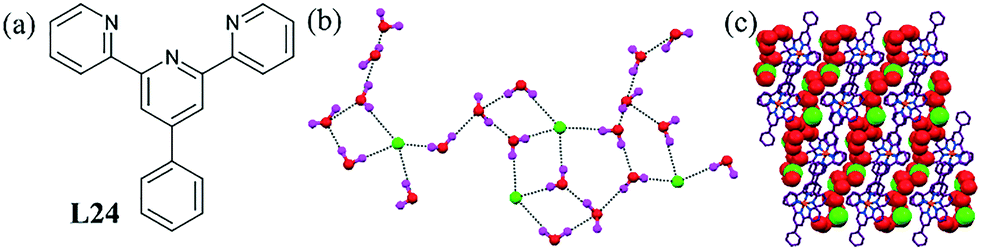 | ||
| Fig. 23 (a) Molecular structure of L24. (b) H-Bonded network of the cluster [Cl4–(H2O)20]4−. (c) Intercalation of the cluster in the complex of L24. Reprinted (adapted) with permission from ref. 64. Copyright 2008 American Chemical Society (CCDC reference number 686054). | ||
Another metal complex of the tripyridine ligand L25 helped to form hydrophilic layers of a chloride–water 2D-network within hydrophobic layers.65 The centro-symmetric repeating 2D-network formed the hydrophilic regions (Fig. 24b). Two pentamers formed a fused system by sharing a chloride ion and water molecule. Such two-fused cyclic pentamers were connected to each other in an inverted manner to form a crown-shaped 8-membered cluster. Overall, the non-bonded O⋯O distances were in the range 2.77(1)–3.03(1) Å and the O⋯Cl distances were in the range 2.91(1)–3.27(1) Å. Two more isostructural metal complexes were also shown to host an almost similar type of chloride–water assembly.
 | ||
| Fig. 24 (a) Molecular structure of L25. (b) H-Bonded 2D chloride–water network. (c) View of the hydrophilic and hydrophobic layers. Reprinted from ref. 65, with permission from Elsevier (CCDC reference number 653942). | ||
The palladium complex of the terpyridine ligand L26 was shown to form a H-bonded chloride–water cluster.66 The cluster contained three non-coordinated water molecules and one Cl− ion and generated a sheet structure. A pure-water tetramer and chloride–water tetramer forming the motif [Cl2–(H2O)6]2− was the minimum repeating unit of the infinite sheet (Fig. 25b). This tetramer formed an infinite chain through an alternating arrangement. The chains were also connected to each other at the corners by water molecules, which produced another fused cyclic octamer ring (Fig. 25c).
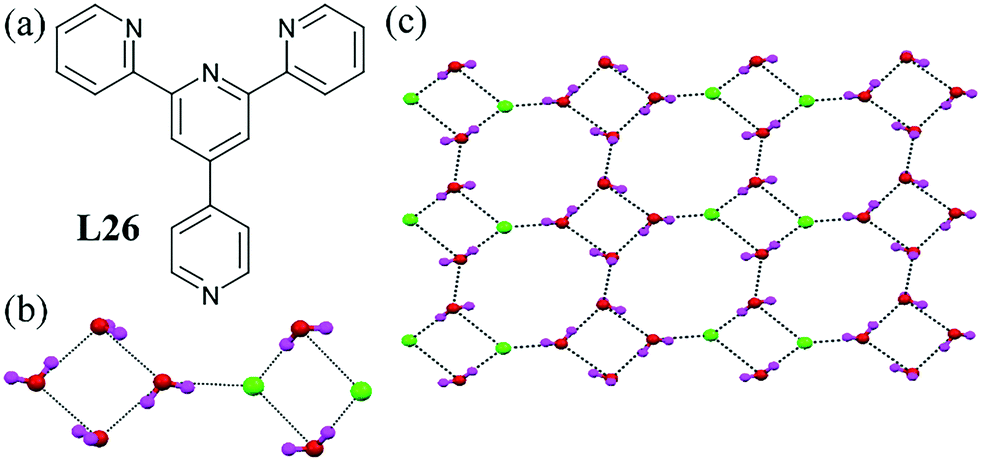 | ||
| Fig. 25 (a) Molecular structure of L26. (b) Minimum repeating unit of the chloride–water cluster [Cl2–(H2O)6]2−. (c) H-Bonded network of the chloride–water cluster. Reprinted from ref. 66, with permission from the Royal Society of Chemistry (CCDC reference number 651576). | ||
The octaaza cryptand L10 (ligand structure described in Fig. 8a) exhibited an intriguing phenomenon of cavity-induced “reductive encapsulation” that occurred upon the complexation of HgCl2 and the formation of a polydodecameric water–chloride cluster [Cl2–(H2O)12]2−.67 All 12 crystallized water molecules were involved in the formation of the dodecameric chloride–water cluster as a repeating unit (Fig. 26b). The decameric core cage was connected with two other core cages by chloride bridging (Fig. 26c). The arrangement within a decameric cage can be described in terms of two semi-fused pentagons. Two pentagons meet to form a chair-like hexamer. This finding is an example of the highest number of water molecules per chloride in either a discrete or an infinite water–chloride cluster.
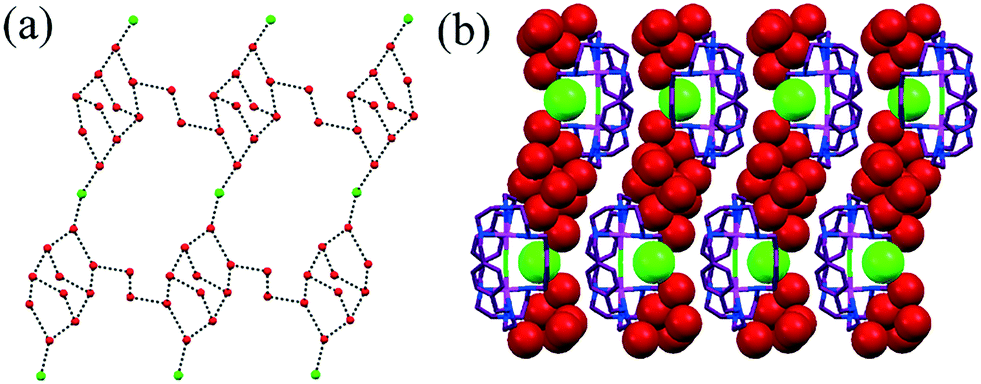 | ||
| Fig. 26 (a) H-Bonded infinite dodecameric water–chloride cluster [Cl2–(H2O)12]2− in L10. (b) Packing view showing progress of the infinite chain through the organic host. Reproduced from ref. 67, with permission from the Royal Society of Chemistry (CCDC reference number 785896). | ||
2.3 Hydrated bromide anion
An unanticipated bromide–water cluster encapsulated in the thiophine-based cryptand L27 has been reported.68 Two bridgehead nitrogen atoms were on the crystallographic C3 axis and one bromide atom was located inside the cavity lying on that axis (Fig. 27b). As shown in Fig. 29c, the encapsulated bromide was hexacoordinated with three “water tetramers”, forming a propeller-shaped hydrate as [Br–(H2O)12]−. The central bromide ion was coordinated with three pairs of water molecules (Fig. 27c). The formation of a discrete, ordered bromide–water cluster with the cryptand was verified by density functional theory (DFT) calculations performed on both the cryptand-[Br–(H2O)12]− complex and the isolated bromide–water cluster. These calculations showed the formation of the stable [Br–(H2O)12]− species without cryptand undergoing a significant re-arrangement upon geometry optimization. The thermal stability of the bromide–water cluster was also established from thermogravimetric analysis and showed the release of water molecules at a temperature of more than 200 °C.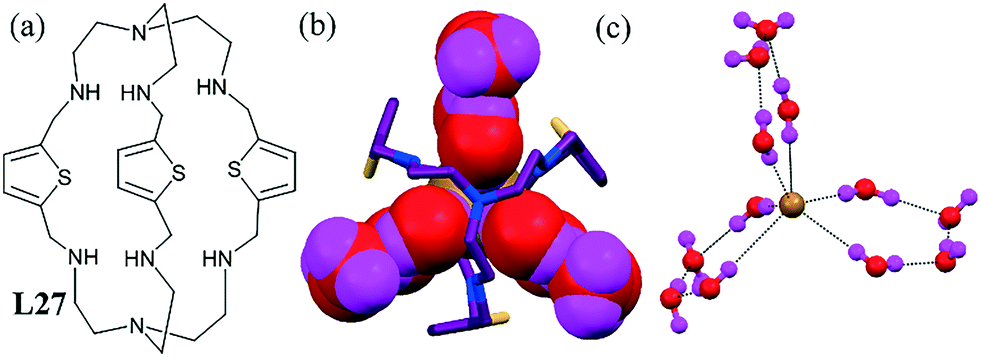 | ||
| Fig. 27 (a) Molecular structure of L27. (b) View showing the bromide–water [Br–(H2O)12]− cluster in the C3 axis mode. (c) A clear view of the bromide–water cluster. Reproduced from ref. 68, with permission from the Royal Society of Chemistry (CCDC reference number 864063). | ||
Cucurbiturils L28 was found to stabilize the isostructural dihalide water cluster consisting of two cyclic mono-bromide hydrates and one cyclic water tetramer.69 The bromide–water cluster was formed through a uudd water tetramer and H-bonded to two [Br–(H2O)3]− clusters as proton acceptors (Fig. 28b). Structurally, the anion–water cluster appeared like a “cut-open and flattened” cubane. The average Br⋯O and O⋯O separations were 3.368(3) Å and 2.831(5) Å, respectively.
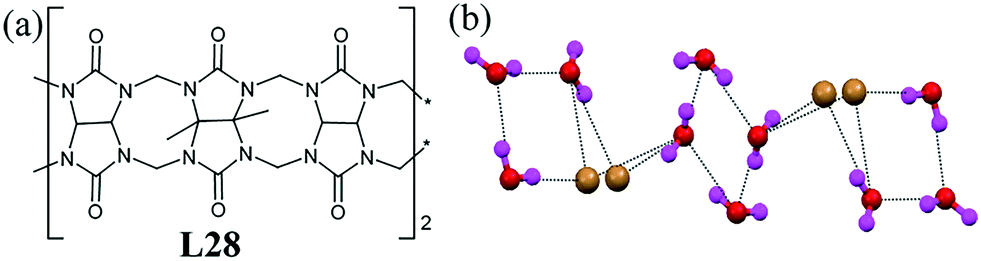 | ||
| Fig. 28 (a) Molecular structure of L28. (b) Structure of the bromide–water cluster in ligand L32. Reprinted (adapted) with permission from ref. 69. Copyright 2012 American Chemical Society (CCDC reference number 902321). | ||
A palladium metal complex of the ligand L29 revealed a very interesting bromide–water assembly of a unique discrete cubane-like dibromide–hexahydrate cluster.70 The bromide ions occupied two opposite corners of the cube (Fig. 29b). The interesting feature of this cluster was that every bromide anion was shared by three water molecules and every water molecule was linked by two bromide anions and two water molecules via H-bonds. One hydrogen of water in the cluster was along each edge of the cube, and each bromide ion was involved in three H-bonding interactions, while each oxygen was connected to another oxygen atom and a bromide ion and accepted one hydrogen from an adjacent oxygen atom on the same edge. The H-bond O⋯O and O⋯Br separations were 2.879(7) Å. The packing diagram, as shown in Fig. 28c, clearly indicates that the water clusters are well-trapped in isolated cavities surrounded by the six cations.
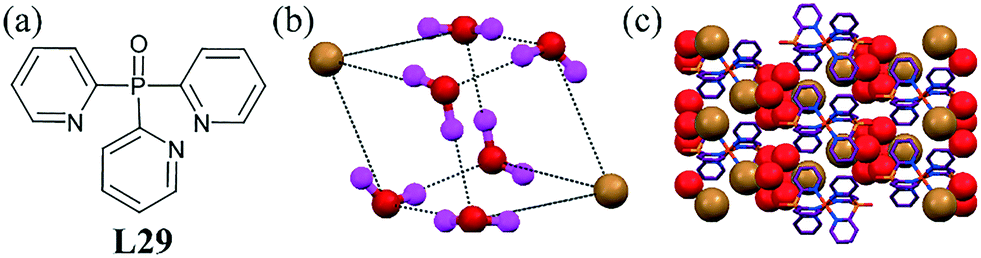 | ||
| Fig. 29 (a) Molecular structure of L29. (b) Cubane-like bromide–water cluster [Br2–(H2O)6]2−. (c) Packing diagram showing the residence of the bromide–water cluster in the crystal system. Reprinted (adapted) with permission from ref. 70. Copyright 2011 American Chemical Society (CCDC reference number 789370). | ||
Another terpyridine system indicated the polymeric nature of the bromide–water assembly, wherein the solid-state structure and DFT calculations suggested that the nature of the anion affected the topology of the halide–water assemblies.71 3D discrete open-cube octameric units could be observed in the crystal structure of protonated L30. These building units were doubly bridged to each other through two bromide ions forming a 1D chain of open-cube octamers (Fig. 30b). The bromide–water cluster was also established by various theoretical calculations.
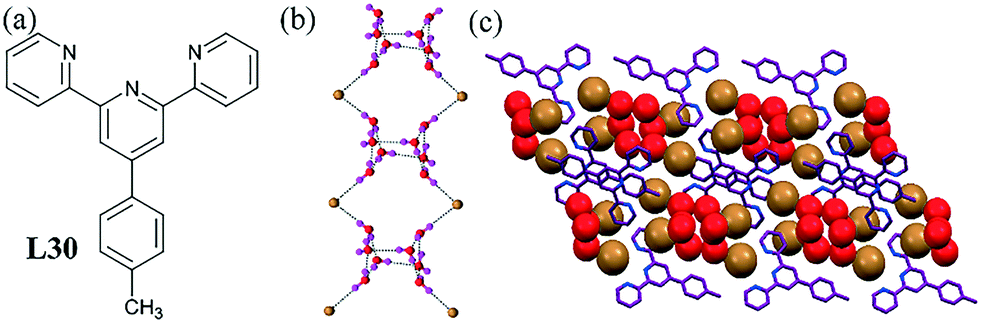 | ||
| Fig. 30 (a) Molecular structure of L30. (b) An open cubane of water clusters connected to each other by bromide ions forming an infinite chain. (c) Packing diagram showing the bromide–water cluster. Reprinted (adapted) with permission from ref. 71. Copyright 2009 American Chemical Society (CCDC reference number 69730). | ||
2.4 Hydrated iodide anion
The planar topological structure of an iodide–water cluster [I–(H2O)6]− in a 3D supramolecular metal complex of ligand L31 was reported.72 In the cluster, the I− anion and water molecules were located in a plane, with each anion connected to two water molecules. Six water molecules in the cluster were divided into two parts: one water monomer and the other a water pentamer. They did not form any cyclic cluster, rather they built an open chain (Fig. 31b). Interestingly, the coplanar water pentamer was a new conformer, different from the cyclic pentamer water cluster. In the anion–water cluster, the O⋯O and I⋯O distances were 2.77 Å and 3.55 Å, respectively. The anion cluster [I–(H2O)6]− together with the anion [Fe(CN)6]2− acted as a 3D crystal host with the other ruthenium complex as the guest molecule, occupying the vacancies of the 3D host framework (Fig. 31c). | ||
| Fig. 31 (a) Molecular structure of L31. (b) Perspective diagram of the [I–(H2O)6]− anion cluster. (c) Packing diagram showing the ruthenium complex surrounded by iodide–water clusters. Reprinted (adapted) with permission from ref. 72. Copyright 2006 American Chemical Society (CCDC reference number 255784). | ||
3. Anion–water cluster of planar anion
Hydrated nitrate anion
A calixarene-based ligand L32 could initiate the formation of a four-clawed crown-like nitrate–water cluster [(NO3)6–(H2O)6]6−.73 The centrosymmetric nitrate–water cluster was composed of six water molecules and two NO3− anions linked by eight O–H⋯O H-bonds (Fig. 32b). The crown-like nitrate–water cluster was incorporated in the inter-space between the layers of cationic ligands. The electrostatic interaction among the nitrate anions and positively charged NMe3 played an important role in stabilizing the cluster. The O⋯O distance within the anionic nitrate–water cluster ranged from 2.738(5) Å to 2.825(5) Å.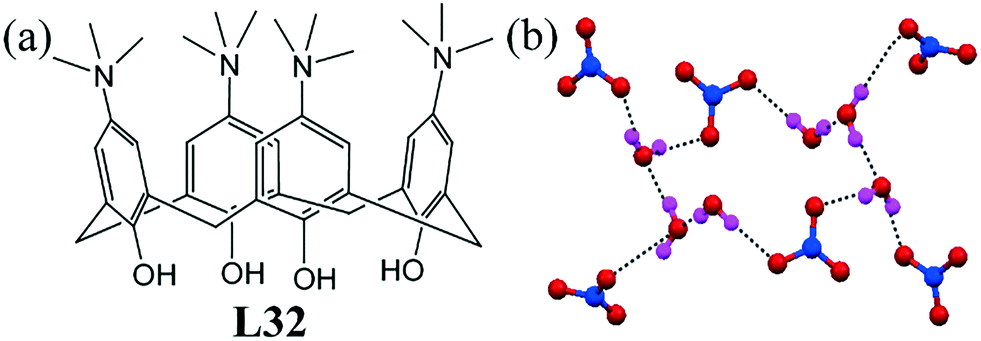 | ||
| Fig. 32 (a) Molecular structure of L32. (b) View of the four-clawed crown-like nitrate–water cluster [(NO3)6–(H2O)6]6−. Reproduced from ref. 73, with permission from the Royal Society of Chemistry (CCDC reference number 818931). | ||
Isomeric abn (aminobenzonitrile) guest-dependent nitrate–water assemblies were shown by ligand L33 during coordination with silver cations.74 Three weakly coordinative abn isomers generated three different types of coordination polymers with the silver(I)–bipyridine system. Nitrate–water clusters, such as [(NO3)4–(H2O)6]∞4−, [(NO3)2–(H2O)2]∞2− and [(NO3)2–H2O]2−, were captured by each polymer, depending on the nature of the abn guest. In the presence of the ortho isomer, the nitrate–water aggregation consisted of four NO3 anions and six H2O molecules, which were arranged to form a 1D hydrogen-bonded chain (Fig. 33b). This chain contained bicyclic [(NO3)3–(H2O)5] subunits and [NO3–H2O] bridges. When the o-abn was replaced by m-abn, it was observed that the new framework captured different nitrate–water aggregations. In this case, an infinite zigzag [(NO3)2–(H2O)2]∞2− chain was observed with nitrate and water alternately arranged (Fig. 33c). The water molecules acted as double H-bond donors towards nitrate anions to form a zigzag chain. The change from m-abn to p-abn resulted in the formation of a discrete H-bonded trimer comprised of two NO3− and one H2O (Fig. 33d). The water molecule acting as a double H-bond donor could bridge two NO3− anions. By changing the isomeric guest molecules, fine tuning of the nitrate–water aggregation was achieved.
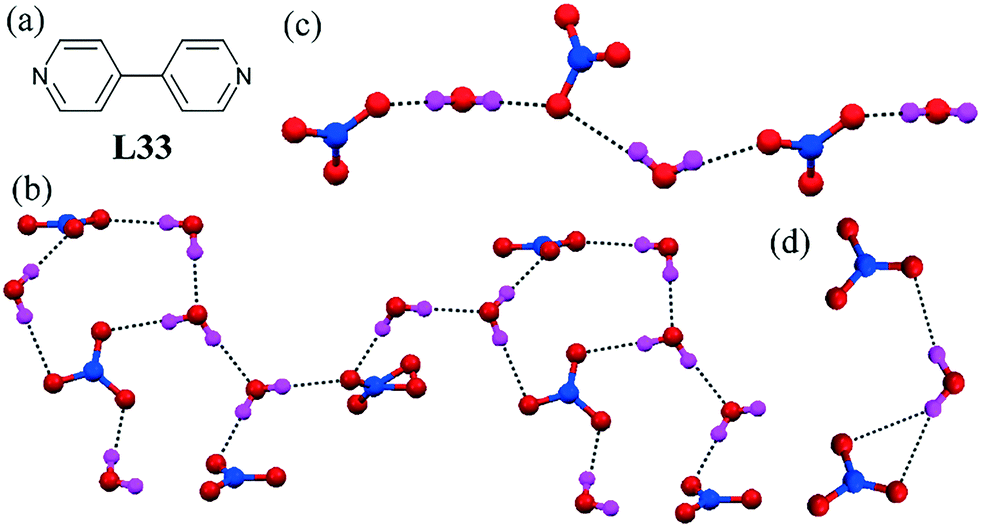 | ||
| Fig. 33 (a) Molecular structure of L33. (b) Ball-and-stick representation of the 1D hydrogen-bonded [(NO3)4–(H2O)6]∞4− chain. (c) Representation of the 1D [(NO3)2–(H2O)2]∞2− zigzag chain. (d) Representation of the discrete trimer of [(NO3)2–H2O]2−. Reproduced from ref. 74, with permission from the Royal Society of Chemistry (CCDC reference numbers 886792–886794). | ||
A 3D coordination polymer of ligand L34 could encapsulate the uncommon nitrate–water cluster [(NO3)4–(H2O)6]4− within its cages.75 A large cage within the crystal was occupied by the four nitrate anions and six water molecules. The striking feature of this nitrate–water cluster was that every NO3− ion was connected with three H2O molecules and every H2O molecule was linked by two NO3− ions (Fig. 34b). The arrangement of nitrate and water molecule was in such a fashion that the cluster had three C2 axes going through the O atoms of water molecules and four C3 axes going through the N atoms of the nitrates.
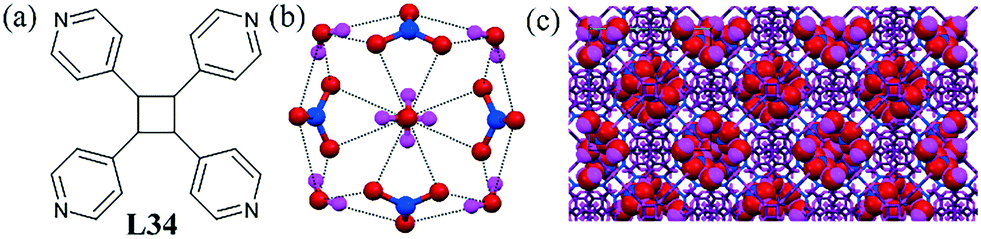 | ||
| Fig. 34 (a) Molecular structure of L34. (b) View of the nitrate–water cluster [(NO3)4–(H2O)6]4−. (c) Packing diagram showing the nitrate–water cluster in the void of the complex. Reprinted (adapted) with permission from ref. 75. Copyright 2009 American Chemical Society (CCDC reference number 704539). | ||
4. Anion–water cluster of tetrahedral anions
4.1 Hydrated sulfate anion
A pyridyl-amide derivative L35 could render the formation of infinite [SO4–(H2O)2]∞2− chains.76 The ligand in the protonated form crystallized with sulfate anions along with two water molecules, with water molecules acting as bridging H-bond donors between the two sulfate anions, leading to the formation of a linear [SO4–(H2O)2]∞2− cluster containing a tetrameric ring of two sulfate anions and two water molecules (Fig. 35b).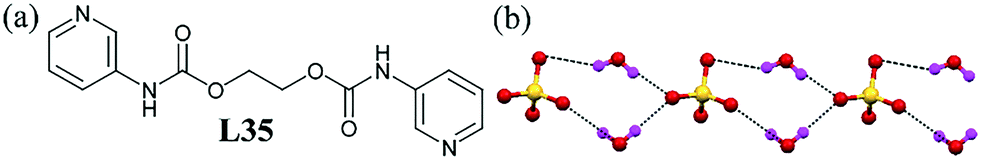 | ||
| Fig. 35 (a) Molecular structure of L35. (b) Illustration of the H-bonded linear chain of the sulfate–water cluster [SO4–(H2O)2]∞2− as a minimum repeating unit. Reproduced from ref. 76, with permission from the Royal Society of Chemistry (CCDC reference number 722508). | ||
A simple ortho-phynylene diamine L36 exhibited the ability to form a helical structure of sulfate anions (Fig. 36c) and a flip-flop water tetramer connected to this sulfate helix.77 All three water molecules interacted with each other forming an extended water structure, which could be described as each water tetramer progressing along the chain perpendicular to the next tetramer (Fig. 36b). This resulted in the formation of sulfate anion helices through noncovalent non-hydrogen-bonded O⋯O contacts.
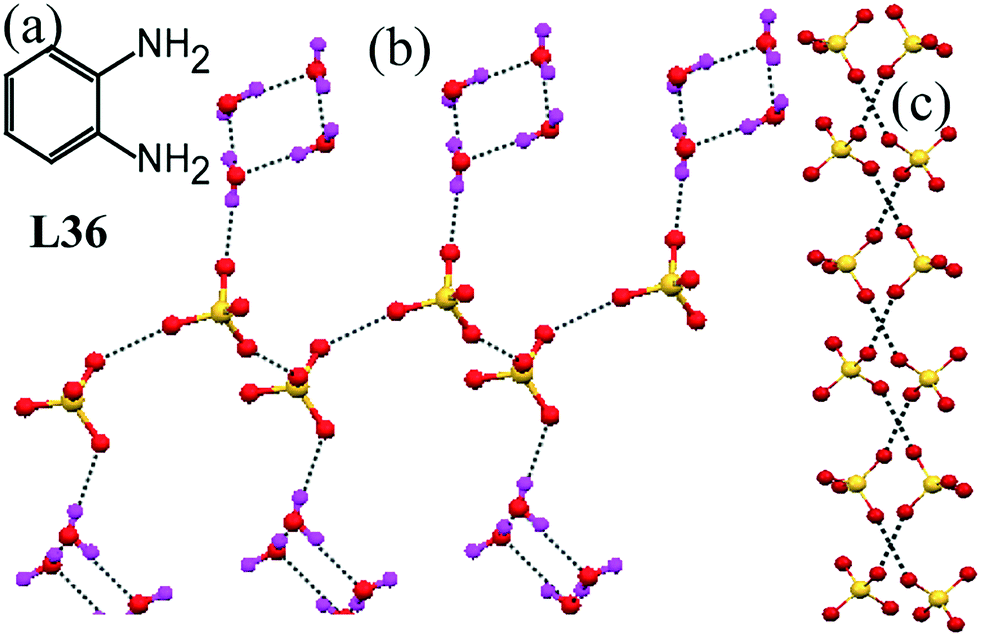 | ||
| Fig. 36 (a) Molecular structure of L36. (b) A sulfate helix showing that each sulfate anion is H-bonded to a water tetramer. (c) Picture showing sulfate helices made by O⋯O interactions. Reproduced from ref. 77, with permission from the Royal Society of Chemistry (CCDC reference number 293163). | ||
Most recently, we reported that tripodal triamine L8 (ligand structure described in Fig. 6a) also formed a discrete sulfate–water tetramer.78 Structural insights into the anion–water clusters indicated that two sulfate ions accepted two hydrogen atoms from two water molecules and formed a discrete sulfate–water tetramer [(SO4)2–(H2O)2]4 (Fig. 37). The sulfate–water tetramer was surrounded by –NH3 and stabilized by electrostatic and H-bonds from the ligand.
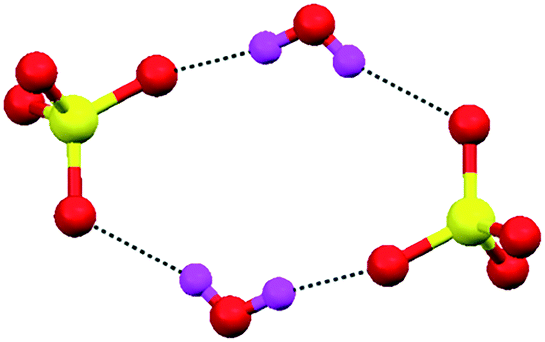 | ||
| Fig. 37 Illustration of the sulfate–water tetramer [(SO4)2–(H2O)2]4− formed by the cationic ligand L8. Reprinted (adapted) with permission from ref. 78. Copyright 2014 American Chemical Society (CCDC reference number 961086). | ||
A bis(guanidinium) ligand L37 led to the formation of a 1D sulfate–water cluster [SO4–(H2O)5]∞2− during sulfate separation from aqueous solutions (Fig. 38b).79 Each sulfate anion in the cluster accepted eight hydrogen bonds from neighboring water molecules, with the range of O⋯O contacts being 2.7 Å to 2.9 Å. The water molecule acted as a bridge among the sulfate anions. The NH hydrogen of the ligand also strongly interacted with the sulfate–water cluster. The peripheral water molecules in the cluster also accepted an additional hydrogen bond from the neighboring stacks, and each sulfate anion accepted three N–H⋯O hydrogen bonds from two ligands from the adjacent stacks.
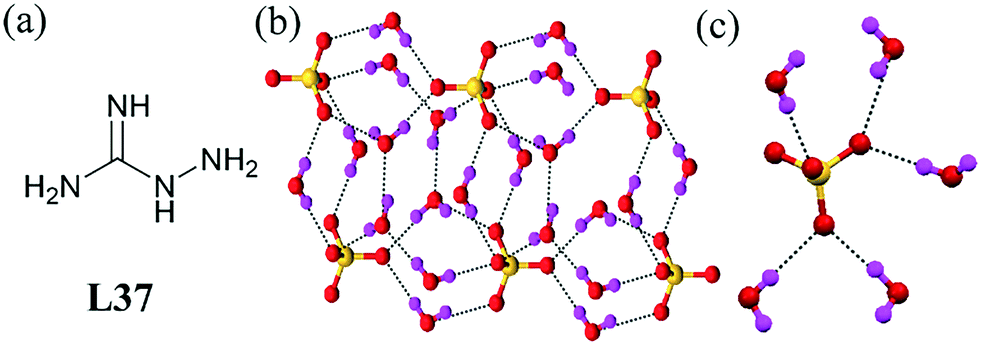 | ||
| Fig. 38 (a) Molecular structure of L37. (b) Hydrogen-bonded 1D chain of the sulfate–water cluster. (c) Repeating unit of the cluster. Reproduced with permission from ref. 79. Copyright Wiley-VCH Verlag GmbH & Co. KGaA (CCDC reference number 1404313). | ||
Recently, we reported the recognition of a hydrated sulfate anion by a longer tripodal amine L38.80 The ligand possessed a large enough cavity to encapsulate a hydrogen sulfate dimer. The encapsulated dimer [HSO4−]2, two exterior HSO4− ions and the water molecules took part in H-bonding assembly and generated an interesting sulfate–water cluster [(HSO4)8–(H2O)4]8−. Two sets of sulfate dimers (encapsulated dimer and exterior dimer) were bridged by a water molecule and formed a sulfate–water pentamer (Fig. 39b). Two such sulfate–water pentamers were finally connected by another water molecule forming a big discrete sulfate–water cluster. Very interestingly, this cluster was connected with the unimolecular capsule through a water molecule at both its ends and this helped to build capsular assemblies of the protonated tripodal ligand (Fig. 39c).
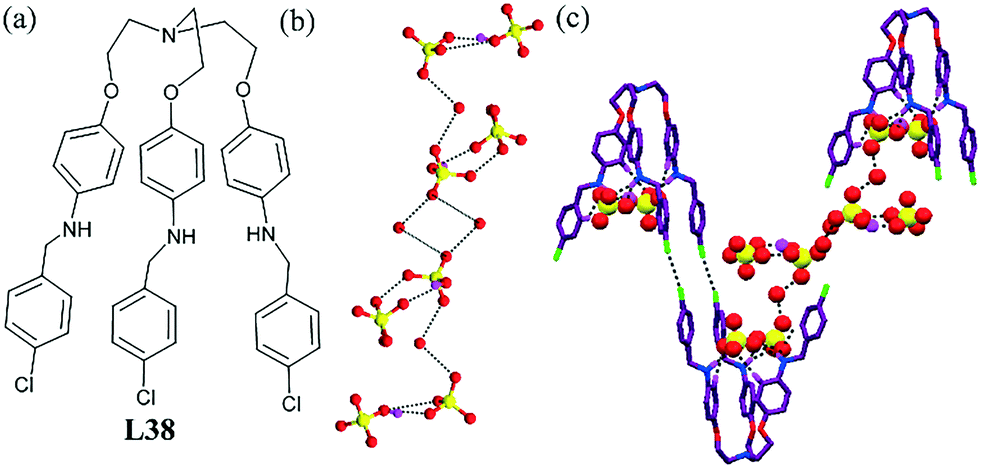 | ||
| Fig. 39 (a) Molecular structure of L38. (b) The big discrete anion–water cluster [(HSO4)8–(H2O)4]8−. (c) Hydrogen sulfate–water cluster [(HSO4)8–(H2O)4]8− and the Cl⋯Cl halogen-bond-assisted supramolecular assembly of a unimolecular capsule along the c-axis. Reprinted from ref. 80 with permission from Taylor & Francis (CCDC reference number 1061890). | ||
A derivative of ligand L39 provided another effective way to separate sulfate anions using the crystallization of hydrated sulfate anions with the help of planar L39.81 The detailed H-bonding interaction in the sulfate–water revealed that sulfate anions were linked together by four water molecules in a centrosymmetric manner giving rise to [(SO4)2–(H2O)4]4− (Fig. 40b). Each sulfate anion in the cluster accepted four H-bonds from water molecules. The sulfate–water cluster [(SO4)2–(H2O)4]4− in the crystal was surrounded by four cationic ligands. Sulfate anions could be successfully separated using crystal engineering techniques by forming a sulfate–water cluster in the solid state.
 | ||
| Fig. 40 (a) Molecular structure of L39. (b) Representation showing a discrete sulfate–water cluster [(SO4)2−(H2O)4]4−. Reproduced with permission from ref. 81. Copyright Wiley-VCH Verlag GmbH & Co. KGaA (CCDC reference number 1430158). | ||
A tripodal tris-urea-based ligand L40 could encapsulate a sulfate–water adduct in its neutral state.82 Binding of the sulfate anions in the solid state showed that the ligand adopted a suitable C3 conformation to encapsulate the sulfate anion and that the urea moiety was involved in H-bonding with the O atoms of the sulfate anion (Fig. 41b). The water molecules acted as a bridge between two encapsulated sulfate anions and produced an interesting rugby-ball-shaped [SO42−–(H2O)3–SO42−] adduct (Fig. 41c). Two hydrogens of each water molecule were involved in H-bonding with the sulfate anion and urea hydrogen also helping stabilization of this discrete sulfate–water in the capsular complex.
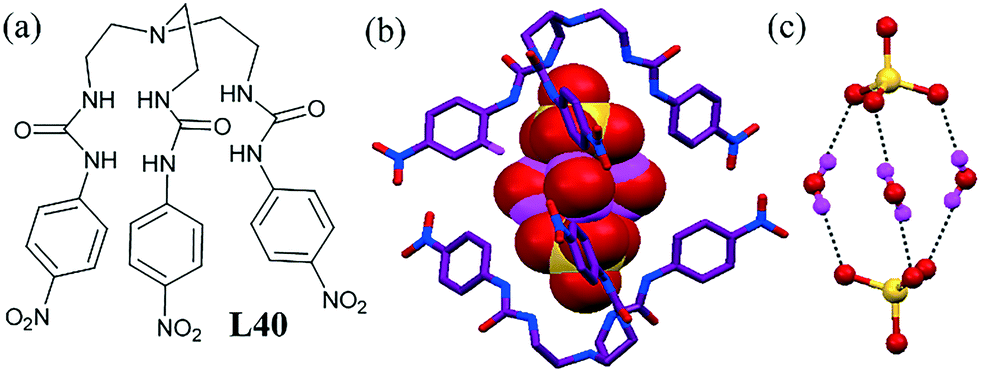 | ||
| Fig. 41 (a) Molecular structure of L40. (b) Space-filling model of the [SO4–(H2O)3–SO4]2− cluster trapped inside the cavity of L40. (c) Illustration displaying the cluster H-bonding in sulfate–water. Reprinted (adapted) with permission from ref. 82. Copyright 2007 American Chemical Society (CCDC reference numbers 623880). | ||
A bowl-shaped metal–organic coordination polymer originating from a cyanuric-acid-platform-based urea ligand L41 formed an unusual sulfate–water cluster [(SO4)4–(H2O)12]8−.83 The tripodal urea ligand on complexation with ZnSO4 initiated the simultaneous encapsulation of as many as four SO42− anions in a supramolecular assembly. Each of the four encapsulated SO42− anions were interconnected by bridging water molecules. Such extensive H-bonding generated a hydrated sulfate cluster [(SO4)4–(H2O)12]8−. In the crystal structure, the cluster was in close contact with the hexa-coordinated Zn2+ ion as [Zn(H2O)6]2+ through two bridging water molecules. Three oxygens of each sulfate anion were coordinated with a water molecule. Such three hydrated sulfate anions around a hexahydrate Zn2+ cation led to the formation of a gigantic sulfate–water cluster (Fig. 42b).
 | ||
| Fig. 42 (a) Molecular structure of L41. (b) Close up view of the [Zn(H2O)6]2+-capped hydrated sulfate anon cluster [(SO4)4–(H2O)12]8−. Reproduced from ref. 83, with permission from the Royal Society of Chemistry (CCDC reference number 1024314). | ||
4.2 Hydrated phosphate anion
A series of reduced Schiff bases L42–L44 functionalized with pyridyl groups rendered the formation of dihydrogen phosphate–water assemblies.84 The assemblies were decorated with H-bond donors and H-bond acceptors, ideal for anion binding. The ligand L42 formed a dihydrogen phosphate–water tetrameric cluster [(H2PO4)2–(H2O)2]2− (Fig. 43b). Crystal analysis indicated that L43 on treatment with TBA–H2PO4 formed a dihydrogen phosphate bridge dimer. The water molecule acted as a bridge between two dihydrogen phosphate dimers [(H2PO4)2–(H2O)2]∞2− and formed a 1D chain of phosphate–water (Fig. 43c). L44 also formed an almost similar phosphate–water cluster as in L43. (Fig. 43b). The oxygen atom of the H2PO4− anion acted as a H-bond donor while the water molecule acted as a H-bond acceptor. Furthermore, H2PO4− anions formed dimers through intermolecular H-bonding interactions, where the oxygen atom of one H2PO4− anion acted as a H-bond donor and another oxygen atom from another H2PO4− anion, which acted as a H-bond acceptor, and vice versa to form a 1D dihydrogen phosphate–water polymer.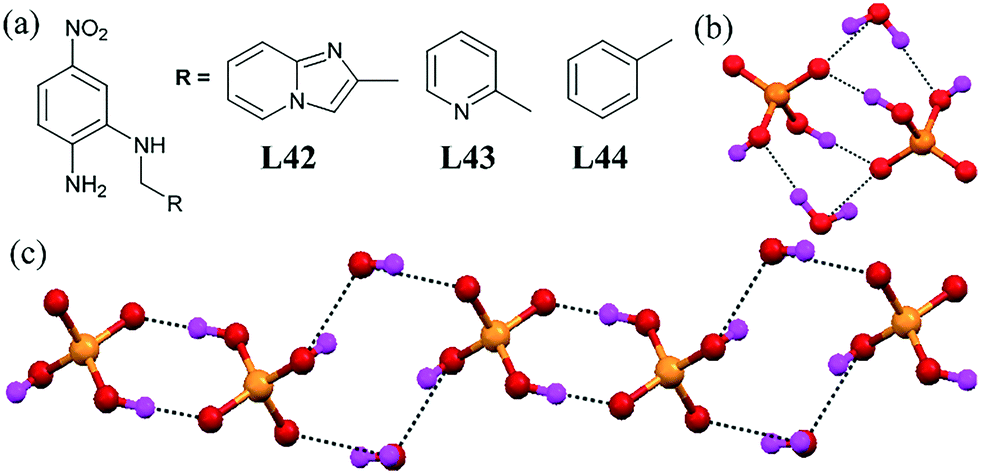 | ||
| Fig. 43 (a) Molecular structures of L42, L43 and L44. (b) Picture showing a discreet dihydrogen phosphate–water tetramer cluster [(H2PO4)2–(H2O)2]2−. (c) 1D dihydrogen phosphate–water cluster containing a phosphate–water tetramer and phosphate dimer. Reproduced from ref. 84, with permission from the Royal Society of Chemistry (CCDC reference numbers 867417 and 867101). | ||
Another type of 1D monohydrogen phosphate–water chain was reported based on the naphthalimide–based ligand L45.85 The HPO42− anion and water molecule underwent self-association to generate a 1D hydrogen phosphate–water chain. The HPO4− anions formed a water-bridged cyclic motif (Fig. 44b). Each of the HPO42− anions interacted with another HPO42− anion through two O–H⋯O interactions to form a hydrogen phosphate dimer. Two such dimers were connected by a water bridge, thus completing the 1D infinite chain.
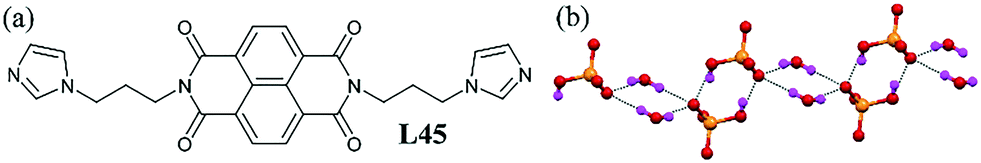 | ||
| Fig. 44 (a) Molecular structure of L45. (b) 1D dihydrogen phosphate–water cluster containing a phosphate–water tetramer and phosphate dimer. Reproduced from ref. 85, with permission from the Centre National de la Recherche Scientifique (CNRS) and the Royal Society of Chemistry (CCDC reference number 911763). | ||
Isomeric pyridine-ethylenediamine-functionalized ligands L46–L48 showed very interesting phosphate–water assembly.86 The protonated crystal structure suggested that the H2PO4− anions and water molecules in the three salts were interconnected with each other through hydrogen bonds to form a layer motif. In the presence of the ligand L46, each adjacent H2PO4− anion interacted with each other to generate chain structures and a similar chain was also formed only by water molecules. Two such chains were connected with each other through the hydrogen bonds between these layers to form a continuous repeat of octameric dihydrogen phosphate–water clusters (Fig. 46b). In the case of ligand L47, two H2PO4− anions were connected by two water molecules to form a dihydrogen phosphate–water tetramer [(H2PO4)2–(H2O)2]∞4− (Fig. 45c). Such a tetramer forms infinite 1D chains through the phosphate anions. As in the case of L46, the H2PO4− anions in L47 also interacted with each other through O⋯H–O hydrogen bonds to form chain structures along the c-axis. In L48, a water molecule and two H2PO4− anions formed a trimeric ring. A dihydrogen phosphate dimer was also connected to this trimeric ring, resulting in the formation of an infinite dihydrogen phosphate–water chain.
 | ||
| Fig. 45 (a) Molecular structures of L46, L47 and L48. (b), (c) and (d) The infinite dihydrogen phosphate–water cluster in L46, L47 and L48, respectively. Reprinted from ref. 86, with permission from Elsevier (CCDC reference numbers 1427889, 1427890 and 1427891). | ||
In one of our earlier studies, we reported the formation of a 1D phosphate–water chain in the presence of the pyridine–urea ligand L49.87 The dihydrogen phosphate complex of L49 revealed extension of the H-bonding contacts on H2PO4− anions, where one water molecule (O1w) acts as a bridging H-bond donor between two symmetry independent H2PO4− anions and forms an unusual water bridge cyclic trimer [H2PO4–H2O–H2PO4]2−, while two of the same symmetric H2PO4− anions form a cyclic dimer [H2PO4–H2PO4]2−via complementary strong H-bonding interactions (Fig. 46b). These two cyclic ring motifs were arranged in an alternating fashion and ultimately led to the formation of an interesting polymeric assembly of hydrated H2PO4− anions.
 | ||
| Fig. 46 (a) Molecular structure of L49. (b) The alternative arrangement of cyclic trimeric and dimeric rings of hydrated dihydrogen phosphate anions. Reprinted (adapted) with permission from ref. 87. Copyright 2014 American Chemical Society (CCDC reference number 943362). | ||
4.3 Hydrated perchlorate anion
A chair-like discrete perchlorate–water cluster was reported based on a 3d–3f-based MOF system containing a simple formic acid ligand L50.88 The crystal structure of the MOF showed chair-like perchlorate anion–water clusters, [(ClO4)2–(H2O)2]2−, formed by two free water molecules and two perchlorate anions with O⋯O separations in the range of 2.8–3.2 Å (Fig. 47c). The anion–water cluster was captured in the channel of the 3D supramolecular structure.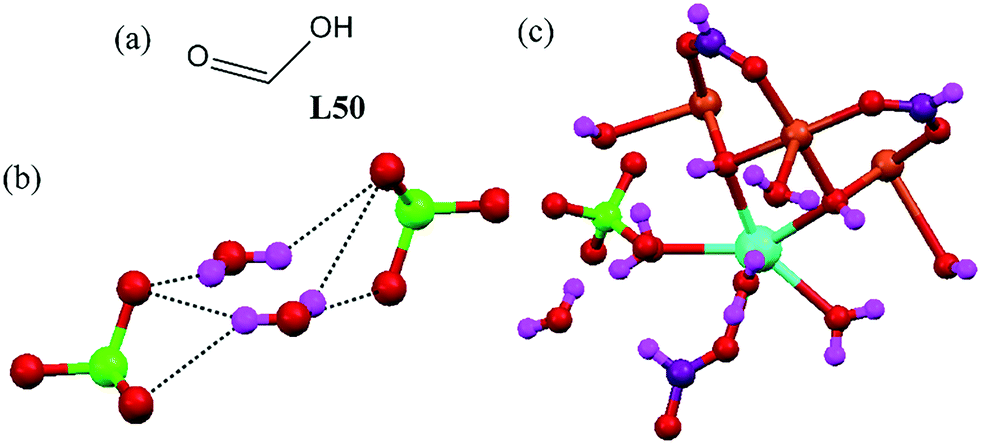 | ||
| Fig. 47 (a) Molecular structure of L50. (b) The discrete chair-like perchlorate–water cluster [(ClO4)2–(H2O)2]2−. (c) The 3d–3f cluster that generated the perchlorate–water cluster. Reproduced from ref. 88, with permission from the Royal Society of Chemistry (CCDC reference number 885435). | ||
5. Hydrated organic anions
The ligand (L2) described in Fig. 2a also formed a bimolecular capsule of an acetate–water cluster.40 The crystal structure showed that the encapsulated acetate ions were bridged by the water molecules, and thus these four water molecules overcame the anion–anion repulsion and held them together inside the dimer. The bridging water molecules were involved in hydrogen bonding interactions to bring other ligands towards the formation of a capsular aggregate. The hexameric cluster [(AcO2)2–(H2O)4]2− was formed from two acetate anions and four water molecules (Fig. 48b). Unlike most other hexameric clusters, the aggregate did not form any chair-like structure. | ||
| Fig. 48 (a) View of the encapsulated acetate ion and water molecules inside the dimeric capsule of ligand L2. (b) Hexameric acetate−water cluster [(AcO2)2–(H2O)4]2−. Reproduced from ref. 40, with permission from the Royal Society of Chemistry (CCDC reference number 733121). | ||
Metal complexes of the ligand L51 with the help of two isomeric bipyridine co-ligands produced a very interesting organic anion–water cluster.89 The cyano-substituted imidazole generated two isomeric organic anion–water aggregations depending on the structure of the bipyridyl co-ligand. In the case of the 2,2′ bipyridyl ligand, a 1D H-bonded imidazole–water aggregation [(imidazole)2–(H2O)4]∞2− containing an octamer water cluster was observed (Fig. 49b), whereas the 4,4′ bipyridyl ligand formed a bag-like binary supramolecular imidazole–water cluster containing two cyano-substituted imidazoles and four water molecules (Fig. 49c). The centrosymmetric octamer water cluster consisted of four water molecules and their centrosymmetric equivalent counterpart. The water octamer was seen as a chair-like hexamer in the core linking two additional water molecules (Fig. 49d). The O⋯O distances in the octamer water cluster were in the range of 2.678(3)–2.774(3) Å, with an average value of 2.729(3) Å. A close inspection of the supramolecular networks formed between the H-bonded water molecules and cyano imidazole anions showed that two imidazole anions and four water molecules were H-bonded to an unprecedented bag-like [(imidazole)2–(H2O)4]∞2− supramolecular cluster, which was further tetrahedrally linked by its four neighbors through the O⋯N hydrogen bond. The two isomeric complexes produced different hydrogen-bonded networks, such as a 1D tape and a 3D 2-fold interpenetrated diamond network, belonging to a pair of isomers.
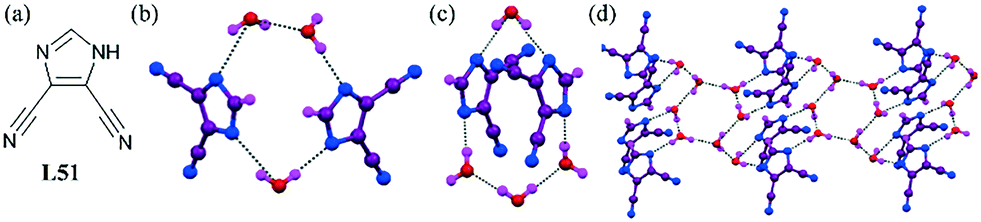 | ||
| Fig. 49 (a) Molecular structure of L51. (b) Cyano-substituted imidazole–water cluster in the presence of 2,2′ bipyridyl. (c) Another [(imidazole)2–(H2O)4]∞2− cluster in the presence of 4,4′ bipyridyl. (d) 1D hydrogen-bonded imidazole–water aggregation. Reproduced from ref. 89, with permission from the Royal Society of Chemistry (CCDC reference numbers 838340 and 838341). | ||
A 3D supramolecular network structure of the cucurbit L52 containing a lengthy pyridinium carboxylate was shown to stabilize the 2D discrete [(pyridinium carboxylate)2–(H2O)10]2− network.90 A detailed crystal structure study suggested that the pyridinium salt was self-assembled with the help of the cucurbit host molecule. The hexyl chain was included inside the cavity, while two pyridyl groups slightly protruded from the portal. The pyridinium carboxylate anion was threaded into the cavity of the cucurbit. The anion and water molecules were self-assembled through hydrogen bonds vertically above the ac layer, forming a 2D infinite network of [(pyridinium carboxylate)2–(H2O)10]2− (Fig. 50b). The anion–water cluster was centrosymmetric and the water cluster in it consisted of three tetramers and two trimers. The average O⋯O distance was 2.84 Å. The anion–water clusters were connected with pseudorotaxanes to form 2D layers.
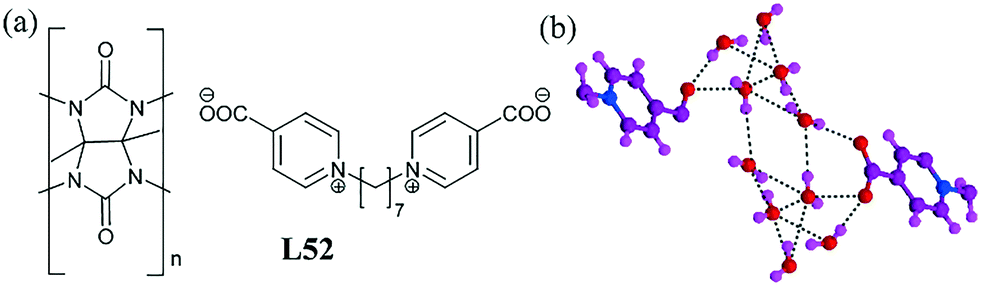 | ||
| Fig. 50 (a) Molecular structure of L52. (b) View showing discrete H-bonded organic anion–water [(pyridinium carboxylate)2–(H2O)10]2−. Reproduced from ref. 90, with permission from the Royal Society of Chemistry (CCDC reference number 705678). | ||
A pyridazine–carboxylic-acid-based ligand L53 exhibited a unique discrete cubane-like carboxylate–water cluster [(COO)2–(H2O)6]2−.91 Structural analysis revealed that the uncoordinated oxygen atom of the carboxylate group acted as a hydrophilic site, which in turn induced the surrounding water molecules to aggregate into a discrete cubane-like anion–water cluster [(COO)2–(H2O)6]2−, where carboxylate anions occupied opposite corners of the water cluster. Every edge of the cube contained one hydrogen atom, and there were two hydrogen atoms outside of the cluster. A detailed structural study indicated that six water molecules formed three H-bonds, whereas the other four water molecules formed two H-bonds (Fig. 51b). The hydrogen-bonded O⋯O distances in the cubane ranged from 2.73 Å to 2.78 Å.
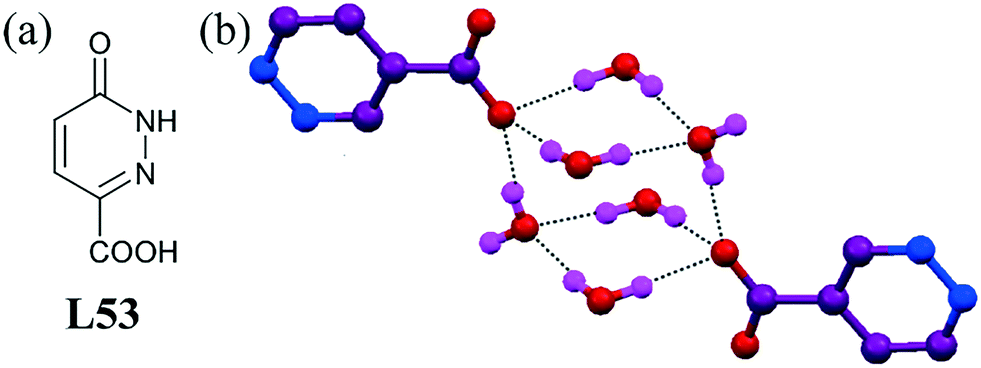 | ||
| Fig. 51 (a) Molecular structure of L53. (b) Structure showing the cubane organic anion–water cluster [(COO)2–(H2O)6]2−. Reprinted from ref. 91, with permission from Elsevier (CCDC reference number 1042389). | ||
A MOF based on ligand L54 helped in the formation of a unique organic anion–water cluster, which was encapsulated into the channels of the cationic host framework.92 The existence of large cationic channels within the crystal structure of the MOF helped in the formation of an unprecedented ladder-like 1D carboxylate–water cluster [(1,3-Hbdc)4–(H2O)9.5]∞4− (Fig. 52b). The (H2O)8 water clusters were enclosed in between these two chains via multiple strong H-bonding interactions between the carboxyl group–water and water–water molecules. Thermogravimetric analysis (TGA) revealed that such a carboxylate–water cluster chain displayed good stability towards heat due to the strong H-bonding interactions among the cationic host framework.
 | ||
| Fig. 52 (a) Molecular structure of L54. (b) View of the ladder-like carboxylate anion–water cluster [(1,3-Hbdc)4–(H2O)9.5]∞4− chain extending along the b axis. Reprinted from ref. 92, with permission from Elsevier (CCDC reference number 763758). | ||
Conclusions
In this review, we attempted to consolidate the solid-state recognition of hydrated anions of varying dimensionality. With the advances in supramolecular chemistry, there have been numerous reviews covering ligand design, binding, variations in physico-chemical properties and the practical application of various anions. Understanding the nature of hydration of the anion is also a fundamental issue. However, due to the unpredictable hydration nature and its implication on the solid-state structure, it is difficult to predict the final outcome of the recognition event. Thus, the volume of literature in this field is relatively lower. Herein, we put in great effort to summarize the widespread structural highlights of hydrated anions, the majority of which are inorganic anions alongside a very few organic anions.As evidenced from the literature survey, fluoride, sulfate and phosphate anions show their desire to be mostly in the hydrated form owing to their small size (fluoride) and/or high charge density (fluoride, sulfate and phosphate) as expected. In all cases, partial hydration of the anions has been observed, which in turn allows these species to interact with the host system possessing complementary H-bonding sites. Understanding the structural aspects of these hydrated anion–host complexes will help us to realize why the recognition of hydrated anions is more important than that of bare anions in the real world. The purpose of this review was to discuss and generalize the various interactions responsible for the recognition of hydrated anion clusters in the solid state. We sub-grouped the receptors based on the functional moiety within the group of a particular anion.
Some striking structural features obtained from the analysis of the crystal structure reported in the literature might draw necessary attention for further development in this field. As an example, the tetrahedron cage formed by L6 was able to encapsulate two isolated pentamers, one of which is only a water pentamer, while the other is a fluoride–water pentamer. This showed the switching of a H-bonding interaction pattern based on the central atom (oxygen vs. fluoride). In the case of the cationic organic matrices of L14, the reversible formation of a chloride–water cluster on dehydration or rehydration may help in controlling the physical properties of the crystalline materials. Hossain et al. showed that a bicyclic ligand (L27) could encapsulate a highly ordered perfect C3 symmetric propeller-shaped bromide–water cluster. This finding represents a step towards the understanding of the complex aqueous environments of an anion within hydrophobic surroundings. In the case of oxyanions, cationic calix[4]arene (L32) stabilizes the planar nitrate anion as a four-clawed crown-like nitrate–water cluster, which offers insight into the nature of the hydration of nitrates in aqueous environments.
Removal and extraction of sulfate or other toxic anionic species from drinking water will be an ideal application of hydrated anion recognition rather than using bare anions. In this regard, an effective approach involving sulfate separation by simple bis(guanidinium) derivatives (L37 and L39) was established by R. Custelcean et al. based on the crystallization of sulfate–water clusters. This has now become an overwhelming trend as an alternative approach for sulfate separation to that of considering the simple bare anion. This greener approach of sulfate separation could inspire an appropriate design of other hosts for the recognition of some important hydrated anions. The conformation-driven self-assembly of hydrogen phosphate and water by ligands L46, L47 and L48 into tape, layer and sandwich-type inorganic–organic hybrid supramolecular networks is very fascinating in the recognition and sensing of phosphate ions. Receptor L51 with water molecules forms an unprecedented bag-like pure organic ion and imidazole–water supramolecular cluster and produces a 3D network with a diamond topology. This result demonstrates an effect of the isomeric host structure (2.2′-pyridyl vs. 4,4′ pyridyl) on the formation of two iso-structural imidazole–water clusters.
The crystal structures described in this highlight illustrate that anions interact with crystallized water molecule(s) and in most of cases they produce a cyclic or closed structure. The plausible intriguing stabilizing factors of anion–water clusters may come from the pre-organized host structure. It may be envisaged that in most of the cases, the host are functionalized with amine, amide, urea/thiourea or pyridine, which are capable of forming various H-bonding and noncovalent interactions. The generation of a desired ordered anion–water cluster is still a difficult task, though it has been observed that a system containing metal ions or an organic molecule with nitrogen (the more basic nitrogen is preferred) atom is prone to support formation of an anion–water cluster. Sometimes the proper design of the receptor (like L6) can also produce some desired anion–water assembly. Future study should address this problem of better understandability and control over the formation of hydrated anions to bring forward some fruitful real-life applications.
Acknowledgements
GD acknowledges the financial support from CSIR and SERB, New Delhi, India. We also thank Central Instrument Facility (CIF) at Indian Institute of Technology (IIT) Guwahati for providing instrument facilities, and DST-FIST for sponsoring a single crystal X-ray diffractometer at Department of Chemistry, IIT Guwahati. N. H. thanks Dr. A. Basu and Dr. S. Dey for inspiring discussion and IIT Guwahati for fellowship.References
- J. W. Steed, Chem. Soc. Rev., 2010, 39, 3686 RSC
.
- V. Amendola, L. Fabbrizzi and L. Mosca, Chem. Soc. Rev., 2010, 39, 3889 RSC
- P. Burkhard, C.-H. Tai, J. N. Jansonius and P. F. Cook, J. Mol. Biol., 2000, 303, 279 CrossRef CAS PubMed
- C.-H. Tai, P. Burkhard, D. Gani, T. Jenn, C. Johnson and P. F. Cook, Biochemistry, 2001, 40, 7446 CrossRef CAS PubMed
- B. T. Burlington and T. S. Widlanski, J. Org. Chem., 2001, 66, 7561 CrossRef
- J. J. He and F. A. Quiocho, Science, 1991, 251, 1479 CAS
- M. Arunachalam and P. Ghosh, Chem. Commun., 2011, 47, 8477 RSC
- P. A. Gale, J. R. Hiscock, C. Z. Jie, M. B. Hursthouse and M. E. Light, Chem. Sci., 2010, 1, 215 RSC
- P. A. Gale, Chem. Commun., 2008, 4525 RSC
- K. A. Schug and W. Lindner, Chem. Rev., 2005, 105, 67 CrossRef CAS PubMed
- I. Ravikumar and P. Ghosh, Chem. Soc. Rev., 2012, 41, 3077 RSC
- R. Custelcean, F. V. Sloop Jr, A. Rajbanshi, S. Wan and B. A. Moyer, Cryst. Growth Des., 2015, 15, 517 CAS
- C. A. Angell, Annu. Rev. Phys. Chem., 1983, 34, 593 CrossRef CAS
- C. A. Angell, R. D. Bressel, M. Gemmati, E. J. Sare and J. C. Tucker, Phys. Chem. Chem. Phys., 2000, 2, 1559 RSC
- A. Chandra, J. Phys. Chem. B, 2003, 107, 3899 CrossRef CAS
- P. Jungwirth and D. J. Tobias, J. Phys. Chem. A, 2002, 106, 379 CrossRef CAS
- K. R. Asmis and D. M. Neumark, Acc. Chem. Res., 2012, 45, 43 CrossRef CAS PubMed
- D. S. Lambrecht, G. N. I. Clark, T. Head-Gordon and M. Head-Gordon, J. Phys. Chem. A, 2011, 115, 11438 CrossRef CAS PubMed
- A. I. Boldyrev and J. Simons, J. Phys. Chem., 1994, 98, 2298 CrossRef CAS
- E. V. Stefanovich, A. I. Boldyrev, T. N. Truong and J. Simons, J. Phys. Chem. B, 1998, 102, 4205 CrossRef CAS
- E. Pluharova, M. Oncak, R. Seidel, C. Schroeder, W. Schroeder, B. Winter, S. E. Bradforth, P. Jungwirth and P. Slavicek, J. Phys. Chem. B, 2012, 116, 13254 CrossRef CAS PubMed
- H. Ohtaki and T. Radnai, Chem. Rev., 1993, 93, 1157 CrossRef CAS
-
D. T. Richens, The chemistry of Aqua Ions, Wiley, Chichester, 1987 Search PubMed
- R. Custelcean, N. J. Williams and C. A. Seipp, Angew. Chem., Int. Ed., 2015, 54, 10525 CrossRef CAS PubMed
- R. Custelcean, N. J. Williams, C. A. Seipp, A. S. Ivanov and V. S. Bryantsev, Chem. – Eur. J., 2016, 22, 1997 CrossRef CAS PubMed
-
Anion Coordination Chemistry, ed. K. Bowman-James, A. Bianchi and E. Garcıa-Espana, Wiley-VCH, Weinheim, 2011 Search PubMed
-
J. L. Sessler, P. A. Gale and W.-S. Cho, Anion Receptor Chemistry: Monographs in Supramolecular Chemistry, RSC Publishing, Cambridge, U.K., 2006 Search PubMed
- N. Gimeno and R. Vilar, Coord. Chem. Rev., 2006, 250, 3161 CrossRef CAS
- R. Custelcean, Chem.
Soc. Rev., 2014, 43, 1813 RSC
- P. Ballester, Chem. Soc. Rev., 2010, 39, 3810 RSC
- M. Wenzel, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2012, 41, 480 RSC
- P. A. Gale, N. Busschaert, C. J. E. Haynes, L. E. Karagiannidis and I. L. Kirby, Chem. Soc. Rev., 2014, 43, 205 RSC
- M. D. Lankshear and P. D. Beer, Acc. Chem. Res., 2007, 40, 657 CrossRef CAS PubMed
- S. O. Kang, J. M. Llinares, V. W. Day and K. Bowman-James, Chem. Soc. Rev., 2010, 39, 3980 RSC
- P. A. Gale, M. E. Light and R. Quesada, Chem. Commun., 2005, 5864 RSC
- D. J. Mercer and S. J. Loeb, Chem. Soc. Rev., 2010, 39, 3612 RSC
- I. Ravikumar and P. Ghosh, Chem. Commun., 2010, 46, 1082 RSC
- M. Cametti and K. Rissanen, Chem. Commun., 2009, 2809 RSC
- S. Chakraborty, R. Dutta, M. Arunachalam and P. Ghosh, Dalton Trans., 2014, 43, 2061 RSC
- M. Arunachalam and P. Ghosh, Chem. Commun., 2009, 5389 RSC
- M. Arunachalam and P. Ghosh, Inorg. Chem., 2010, 49, 943 CrossRef CAS PubMed
- S. Chakraborty, R. Dutta, B. M. Wong and P. Ghosh, RSC Adv., 2014, 4, 62689 RSC
- M. Arunachalam and P. Ghosh, Chem. Commun., 2011, 47, 6269 RSC
- Q.-Q. Wang, V. W. Day and K. Bowman-James, Angew. Chem., Int. Ed., 2012, 51, 2119 CrossRef CAS PubMed
- S. Dalapati, M. A. Alam, R. Saha, S. Janaa and N. Guchhait, CrystEngComm, 2012, 14, 1527 RSC
- M. N. Hoque and G. Das, CrystEngComm, 2014, 16, 4447 RSC
- M. A. Hossain, M. A. Saeed, A. Pramanik, B. M. Wong, S. A. Haque and D. R. Powell, J. Am. Chem. Soc., 2012, 134, 11892 CrossRef CAS PubMed
- J. L. Atwood, L. J. Barbour, T. J. Ness, C. L. Raston and P. L. Raston, J. Am. Chem. Soc., 2001, 123, 7192 CrossRef CAS PubMed
- M. A. Hossain, J. M. Llinares, S. Mason, P. Morehouse, D. Powell and K. Bowman-James, Angew. Chem., Int. Ed., 2002, 41, 2335 CrossRef PubMed
- U. Manna, R. Chutia and G. Das, Cryst. Growth Des., 2016, 16, 2893 CAS
- A. Basu and G. Das, Chem. Commun., 2013, 49, 3997 RSC
- J. R. Butchard, O. J. Curnow, D. J. Garrett and R. G. A. R. Maclagan, Angew. Chem., Int. Ed., 2006, 45, 7550 CrossRef CAS PubMed
- P. S. Lakshminarayanan, E. Suresh and P. Ghosh, Angew. Chem., Int. Ed., 2006, 45, 3807 CrossRef CAS PubMed
- K. K. Bisht, A. C. Kathalikkattil and E. Suresh, Cryst. Growth Des., 2012, 12, 556 CAS
- D.-X. Wang, S.-X. Fa, Y. Liu, B.-Y. Houa and M.-X. Wang, Chem. Commun., 2012, 48, 11458 RSC
- M. N. Hoque, A. Basu and G. Das, Cryst. Growth Des., 2012, 12, 2153 CAS
- R. Saha, S. Biswas, I. M. Steele, K. Dey and G. Mostafa, Dalton Trans., 2011, 40, 3166 RSC
- R. Custelcean and M. G. Gorbunova, J. Am. Chem. Soc., 2005, 127, 16362 CrossRef CAS PubMed
- A. K. Ghosh, D. Ghoshal, J. Ribas, G. Mostafa and N. R. Chaudhuri, Cryst. Growth Des., 2006, 6, 36 CAS
- J. S. Casas, M. D. Couce, A. Sanchez, J. Sordo and E. M. V. Lopez, Inorg. Chem. Commun., 2013, 30, 156 CrossRef CAS
- M. N. Kopylovich, E. A. Tronova, M. Haukka, A. M. Kirillov, V. Y. Kukushkin, J. J. R. Fraústo da Silva and A. J. L. Pombeiro, Eur. J. Inorg. Chem., 2007, 4621 CrossRef CAS
- M. Moustapha-Sow, O. Diouf, M. Gaye, A. Salam-Sall, G. Castro, P. Perez-Lourido, L. Valencia, A. Caneschi and L. Sorace, Cryst. Growth Des., 2013, 13, 4172 CAS
- S. S. Bhat and V. K. Revankar, J. Chem. Crystallogr., 2016, 46, 9 CrossRef CAS
- R. R. Fernandes, A. M. Kirillov, M. F. C. Guedes da Silva, Z. Ma, J. A. L. da Silva, J. J. R. Frausto da Silva and A. J. L. Pombeiro, Cryst. Growth Des., 2008, 8, 782 CAS
- S. K. Padhi, R. Sahu and V. Manivannan, Polyhedron, 2010, 29, 709 CrossRef CAS
- R. Saha, S. Biswas, I. M. Steele, K. Dey and G. Mostafa, Dalton Trans., 2011, 40, 3166 RSC
- A. Pati, J. Athilakshmi, V. Ramkumar and D. K. Chand, CrystEngComm, 2014, 16, 6827 RSC
- M. A. Saeed, A. Pramanik, B. M. Wong, S. A. Haque, D. R. Powell, D. K. Chand and M. A. Hossain, Chem. Commun., 2012, 48, 8631 RSC
- W.-J. Chen, L.-S. Long, R.-B. Huang and L.-S. Zheng, Cryst. Growth Des., 2013, 13, 2507 CAS
- A. Bakhoda, H. R. Khavasi and N. Safari, Cryst. Growth Des., 2011, 11, 933 CAS
- B. Dey, S. R. Choudhury, P. Gamez, A. V. Vargiu, A. Robertazzi, C.-Y. Chen, H. M. Lee, A. D. Jana and S. Mukhopadhyay, J. Phys. Chem. A, 2009, 113, 8626 CrossRef CAS PubMed
- W. Dong, Y. Ou-Yang, H.-B. Song, D.-Z. Liao, Z.-H. Jiang, S.-P. Yan and P. Cheng, Inorg. Chem., 2006, 45, 1168 CrossRef CAS PubMed
- L.-L. Liu, Z.-G. Ren, L.-M. Wan, H.-Y. Dinga and J.-P. Lang, CrystEngComm, 2011, 13, 5718 RSC
- D. Sun, F.-J. Liu, R.-B. Huangb and L.-S. Zheng, CrystEngComm, 2012, 14, 7872 RSC
- D. Liu, H.-X. Li, Z.-G. Ren, Y. Chen, Y. Zhang and J.-P. Lang, Cryst. Growth Des., 2009, 9, 4562 CAS
- Y. Xia, B. Wu, Y. Liu, Z. Yang, X. Huang, L. Hea and X.-J. Yang, CrystEngComm, 2009, 11, 1849 RSC
- P. Raghavaiah, S. Supriya and S.
K. Das, Chem. Commun., 2006, 2762 RSC
- M. N. Hoque and G. Das, Cryst. Growth Des., 2014, 14, 2962 CAS
- R. Custelcean, N. J. Williams and C. A. Seipp, Angew. Chem., Int. Ed., 2015, 54, 10525 CrossRef CAS PubMed
- M. N. Hoque, U. Manna and G. Das, Supramol. Chem., 2016, 28, 284 CrossRef CAS
- N. Custelcean, J. Williams, C. A. Seipp, A. S. Ivanov and V. S. Bryantsev, Chem. – Eur. J., 2016, 22, 1997 CrossRef PubMed
- D. A. Jose, D. K. Kumar, B. Ganguly and A. Das, Inorg. Chem., 2007, 46, 5817 CrossRef CAS PubMed
- R. Dutta, B. Akhuli and P. Ghosh, Dalton Trans., 2015, 44, 15075 RSC
- S. Dalapati, M. A. Alam, S. Janaa and N. Guchhait, CrystEngComm, 2012, 14, 6029 RSC
- J. K. Nath and J. B. Baruah, New J. Chem., 2013, 37, 1509 RSC
- J. Huang, T.-P. Liu, L.-H. Huo, Z.-P. Deng and S. Gao, J. Mol. Struct., 2017, 1127, 361 CrossRef CAS
- M. N. Hoque, A. Basu and G. Das, Cryst. Growth Des., 2014, 14, 6 CAS
- Z.-Y. Li, J.-S. Yang, R.-B. Liu, J.-J. Zhang, S.-Q. Liu, J. Nia and C.-Y. Duan, Dalton Trans., 2012, 41, 13264 RSC
- T. Hu, X. Zhao, X. Hu, Y. Xu, D. Sun and D. Sun, RSC Adv., 2011, 1, 1682 RSC
- X. Sun, B. Li, C. Xia, X. Zhoua and H. Zhang, CrystEngComm, 2012, 14, 8525 RSC
- S. Deng, J. Chen, J. Nie, S. Liu, X. Duan, W. Xiao and N. Zhang, Inorg. Chem. Commun., 2015, 55, 77 CrossRef CAS
- E. Tang, Y.-M. Dai, Y. Chen, J.-P. Lang, Y.-G. Yao and N.-S. Weng, Inorg. Chem. Commun., 2010, 13, 887 CrossRef CAS
Footnote |
| † Present address: Key Laboratory of Bioinorganic and Synthetic Chemistry of Ministry of Education, School of Chemistry, Sun Yat-Sen University, Guangzhou 510275, P. R. China. |
| This journal is © The Royal Society of Chemistry 2017 |