
Structures and applications of metal–organic frameworks featuring metal clusters
Ling
Qin
ab and
He-Gen
Zheng
 *a
*a
aState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Collaborative Innovation Center of Advanced Microstructures, Nanjing University, Nanjing 210093, P.R. China. E-mail: zhenghg@nju.edu.cn; Fax: +86 25 83314502
bDepartment of Chemical Engineering and Food Processing, Xuancheng Campus, Hefei University of Technology, Xuancheng, 242000, Anhui, P.R. China
First published on 2nd December 2016
Abstract
Metal–organic frameworks (MOFs), self-assembled from metal ions or metal clusters with organic ligands, are well known for their intriguing structure, permanent porosity, and tunable properties and have shown great prospect for various applications. In the present review, we mainly highlight recent significant examples of metal clusters in our team and provide a comprehensive review on the structure of metal cluster-based MOFs and their applications including for sensing metal ions, small molecules and gases, nitroaromatics and explosives, as well as many other applications. We classified two different coordination polymers: (i) homometallic coordination polymers (examples of coordination polymers with single and mixed types of secondary building units) and (ii) heterometallic coordination polymers (examples of coordination polymers based on metal–oxygen clusters and W–Cu–S clusters).
 Ling Qin | Dr. Ling Qin received her PhD in Inorganic Chemistry in 2014 from Nanjing University under the supervision of Professor He-Gen Zheng. Then she joined the faculty at the Department of Chemical Engineering and Food Processing, Xuancheng Campus, Hefei University of Technology. Her interests focus on the design and synthesis of metal–organic frameworks with multifunctional properties, including chirality, sensing, gas storage, separation and catalysis. |
 He-Gen Zheng | Prof. He-Gen Zheng received his PhD in inorganic chemistry under the supervision of Prof. Xin-Quan Xin from Nanjing University (1998). He has been a full professor at Nanjing University since 2005. He worked as a visiting scholar at The Hong Kong University of Science and Technology (1997–1998) and had post-doctoral experience at Hokkaido University, Japan (1999–2000). His research interests focus on crystal engineering of coordination, supramolecular chemistry and dye sensitized solar cells. |
Introduction
Metal–organic frameworks (MOFs) have received considerable attention due to their fascinating structures and topologies and potential applications in gas storage, sensors, separation, magnetism, and catalysis.1 The utilization of polynuclear metal clusters as building blocks creates more opportunities to construct highly connected frameworks because of their large surface areas, which enables them to more readily accommodate the steric demands of organic linkers. Furthermore, the coordination polymers constructed with polynuclear cluster units should be more stable, and the large size of the clusters may result in larger pore dimensions relative to those in noncluster frameworks with the same connectivity.2In this highlight review we focus on selected examples of metal cluster-based MOFs and briefly on their applications. We mainly introduce our groups' research work and also introduce some related work of other groups. First, we will focus on MOFs with homometallic clusters. The content mainly includes two aspects: cluster–organic frameworks with single and mixed types of secondary building units. Then, we will highlight studies on heterometallic coordination polymers. The content mainly includes two aspects: coordination polymers based on heterometallic oxygen clusters and W–Cu–S clusters. Finally, we will discuss the relationships between the metal cluster units and the structures; introduction of cluster secondary building units can reduce interpenetration, which provides MOFs with permanent porosity and rigid architectures.
Homometallic coordination polymers
The use of secondary building units (SBUs) instead of single metal ions at the network vertices was a powerful means of directing the final framework topology and structure and has a significant impact on the properties of the resulting MOFs. Among the many types of SBUs, the tetranuclear [Zn4O(COO)6] and dinuclear [Cu2(COO)4] SBUs were widely researched and used, which were synthesized in self-assembly reactions and considered as six-connected octahedral and four-connected tetragonal nodes, respectively.3Cluster–organic frameworks with dinuclear secondary building units
Three new metal–organic frameworks (MOFs) were designed by the assembly of flexible V-shaped ligands and paddle-wheel secondary building units.4 The topology, interpenetration numbers and porosity of frameworks have been well controlled by a solvent system.Since 4,4′-dicarboxydiphenylamine (H2L) is a flexible V-shaped ligand,5 there were two possibilities for constructing the paddle-wheel second building units (Cu2(CO2)4) (Scheme 1): one way was the rctt (type I), and the other way was the rtct (type II).6 The Cu2(CO2)4 units were connected by L2− ligands to generate three different 3D nets. As shown in Fig. 1, by the systematic variation of the solvent, three unprecedented novel MOFs were obtained: {[Cu(L)(DMF)]·5/2H2O}n (1) with three-fold interpenetrated lvt topology; {[Cu2(L)2(H2O)2]·3/2H2O}n (2) with three-fold interpenetrated dia topology; and {[Cu(L)(H2O)]·H2O}n (3) with five-fold interpenetrated lvt topology. This presented an amazing example of control over the topology, interpenetration and porosity of MOFs via solvent by the assembly of flexible V-shaped ligands and paddle-wheel SBUs. The DMF–H2O system favored a three-fold interpenetrated framework with lvt topology; the DMF–CH3CN–H2O system favored a three-fold interpenetrated framework with dia topology; and the CH3CN–H2O system favored a five-fold interpenetrated framework with lvt topology. The porosity of the MOFs was also adjusted. Even though many interpenetrated frameworks with various topologies have been widely reported, controlling frameworks with different topologies, different interpenetrated numbers, and porosity via the regulation of solvents has been still rare.
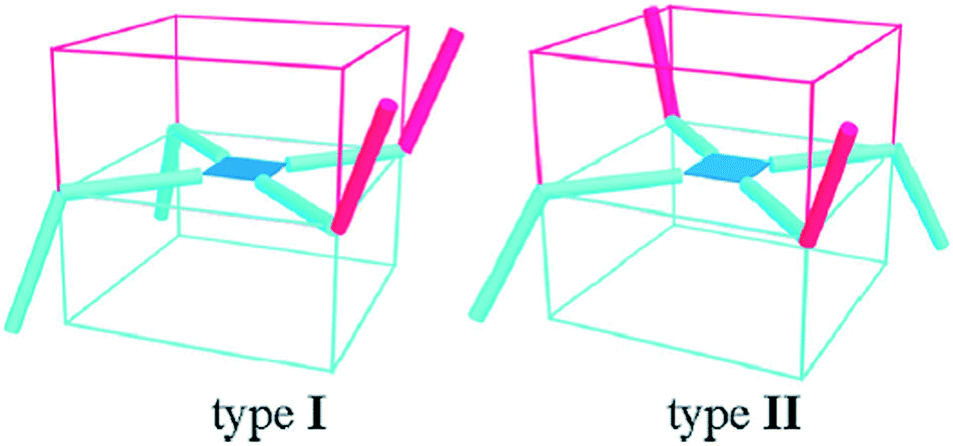 | ||
| Scheme 1 Schematic of two types of the supposed assembly way for the V-shaped ligand (thick rod) and copper paddlewheel SBU (the blue square): type I was the assembly way of up–up–down–down (rctt), and type II was the assembly way of up–down–up–down (rtct). | ||
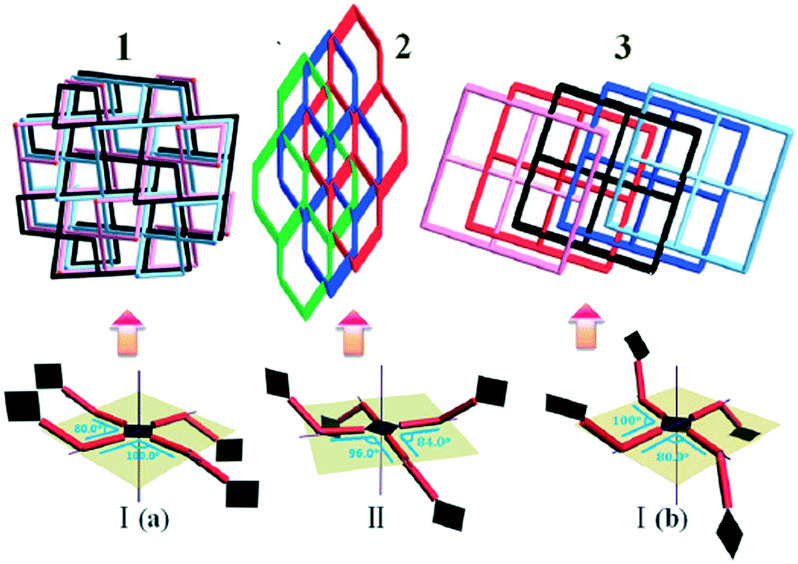 | ||
| Fig. 1 The topologies of 1, 2, and 3: three-fold interpenetrated with lvt net for 1 assembled using the rctt way; three-fold interpenetrated with dia net for 2 assembled using the rtct way; and five-fold interpenetrated with lvt net for 3 assembled using the rctt way. | ||
One dinuclear non-interpenetrated 3D chiral porous framework,7 {[Cd2(TPPBDA)(bpdc)3/2(H2O)2]·1/2(CO3)}n (4), prepared by introducing SBUs constructed by connections of carboxylate and tetratopic pyridyl ligands8 to metal centers has been successfully presented. Each Cd(II) with a distorted octahedral coordination sphere was coordinated by two nitrogen atoms of TPPBDA, three carboxyl oxygen atoms, and one water molecule. Two adjacent Cd cations were linked in sequence by the carboxyl groups of bpdc2−, forming an infinite rod-shaped secondary building unit. Each rod-shaped SBU linked four adjacent SBUs through the TPPBDA molecules to generate a non-interpenetrated 3D framework. The fluorescence intensities and maximum emission peaks for compound 4 showed certain correlations with the dielectric constant values of the solvents. The correlations were obtained by using separately non-protonic and protonic solvents, as shown in Fig. 2. With the increase of the dielectric constant of the protonic solvent guests, the maximum emission peaks were red-shifted, showing a positive correlation effect for non-protonic solvents. On the contrary, the maximum emission peaks were blue-shifted with the increase of the dielectric constant of the nonprotonic solvent guests, showing a negative correlation effect. To enable a deep discussion on the issues about the emission of the compound, which was dependent on the solvent dielectric constant and the protonic property, we checked whether the TPPBDA ligand itself has the same solvent sensitivity. The results showed that the emission position for the TPPBDA ligand was not related with the dielectric constant of the solvent or whether it was protonic.
 | ||
| Fig. 2 The maximum emission peaks of compound 4 against dielectric constant values of solvents. | ||
In 2015, one luminescent Zn(II) metal–organic framework,9 {[Zn2(L)2(dpyb)]}n (5), with a “paddlewheel” secondary building unit, was prepared from a π-conjugated thiophene-containing carboxylic acid ligand (H2L = 3,3′-(thiophene-2,5-diyl)dibenzoic acid, dpyb = 1,4-di(pyridin-4-yl)benzene). Two crystallographically equivalent Zn(II) cations were bridged by four carboxylate groups which adopted a bis-bidentate coordination mode to form a dinuclear Zn(II) SBU. Such SBUs were bridged by dpyb ligands to form a 2D (4,4)-sql network. Compound 5 suggested a strong photoluminescence and photoluminescence quenching effect with nitroaromatic compounds. Importantly, it exhibited very highly sensitive and selective detection of picric acid compared to other nitroaromatic explosives. The photoluminescence intensities were strongly dependent on the organic solvents; the strongest emissions were shown in DMA. However, the nitrobenzene emulsions displayed a complete quenching effect. Such solvent-dependent luminescence properties inspired us to investigate the possibility of sensing other nitroaromatic explosives. To examine the sensing sensitivity toward nitroaromatic explosives, a series of suspensions in DMA with gradually increasing concentrations of nitroaromatic explosives in DMA solution were prepared to monitor the photoluminescence response. The photoluminescence intensities decreased sharply with the gradual droping of PA solution to DMA emulsion.
The photoluminescence quenching efficiency can be quantitatively explained by the Stern–Volmer (SV) equation, (I0/I) = 1 + Ksv[Q], in which Ksv is the quenching constant (M−1), [Q] is the molar concentration of the nitro analyte, and I0 and I are the photoluminescence intensities before and after addition of the nitro analyte, respectively. The Stern–Volmer plots for PA were nearly linear at low concentrations (R2 = 0.9989) (Fig. 3). The plots subsequently deviated from linearity and bent upwards at higher concentrations. Such phenomena of nonlinear SV plots may be due to self-absorption or an energy-transfer process.10 Compound 5 showed the highest Ksv values with PA, at 2.40 × 104, which is comparable to those of known organic polymers. The quenching constant for PA is ca. 68 times greater than for NB. The high quenching constant and the low detection concentration for PA indicated easy identification of the presence of a trace quantity of explosive. In addition, the MOF 5 also responded rapidly and sensitively to the PA vapor, which was slightly lower than that to the solution. Within 1 min, a quenching maximum percentage of approximately 80% was reached.
 | ||
| Fig. 3 (a) Photoluminescence spectra and (b) SV plot of MOF 5 by gradual addition of 5 mM PA in DMA. The inset shows the emission quenching linearity relationship at low concentrations of PA. | ||
To better understand the high selectivity towards PA, the quenching mechanism was investigated. The absorption band intensities at 296 nm and 295 nm were reduced, and new absorption bands at 363 nm and 362 nm were observed, indicating the formation of charge transfer complexes upon addition of PA. Another possible reason for the quenching may be the resonance energy transfer mechanism.11 As we have assumed, the absorption band of PA had the greatest degree of overlap with the photoluminescence spectra in DMA, while all the other nitro analytes almost had no overlap. As a result, the coexistence of electron transfer and resonance energy transfer made PA exhibit high photoluminescence quenching compared to other nitro analytes.
Cluster–organic frameworks with trinuclear secondary building units
Three isostructural Ln-BTB frameworks (Ln = Eu (6), Dy (7), and Yb (8)) (H3BTC = 1,3,5-benzenetribenzoate) were synthesized and structurally characterized, with mononuclear and trinuclear units as nodes, by B. Zhao.12 Structure analysis suggested that all of these compounds were isostructural and crystallize in the orthorhombic system with space group Pbcm. Thus, compound 6 was selected as a representative example for a detailed description. The structure comprised three parts: two distinct Eu3+ ions and one [Eu3] cluster. Both the eight-coordinated Eu3 and Eu4 had a distorted square antiprism geometry. Eu2, Eu1, and Eu1a were connected in a trinuclear [Eu3] unit by carboxyl groups. Three distinct parts were connected by a BTB3− ligand to form a 3D anionic framework. The luminescence investigations revealed that compound 6 can sensitively and selectively detect Al3+, while compound 7 could not detect Al3+ among various cations. More importantly, 6 as an Al3+ sensor can be reused at least five times, which represented the first recyclable MOF-supported Al3+ sensor. Additionally, magnetic investigations on 7 also were carried out, showing a single-molecule-magnet behavior (Fig. 4).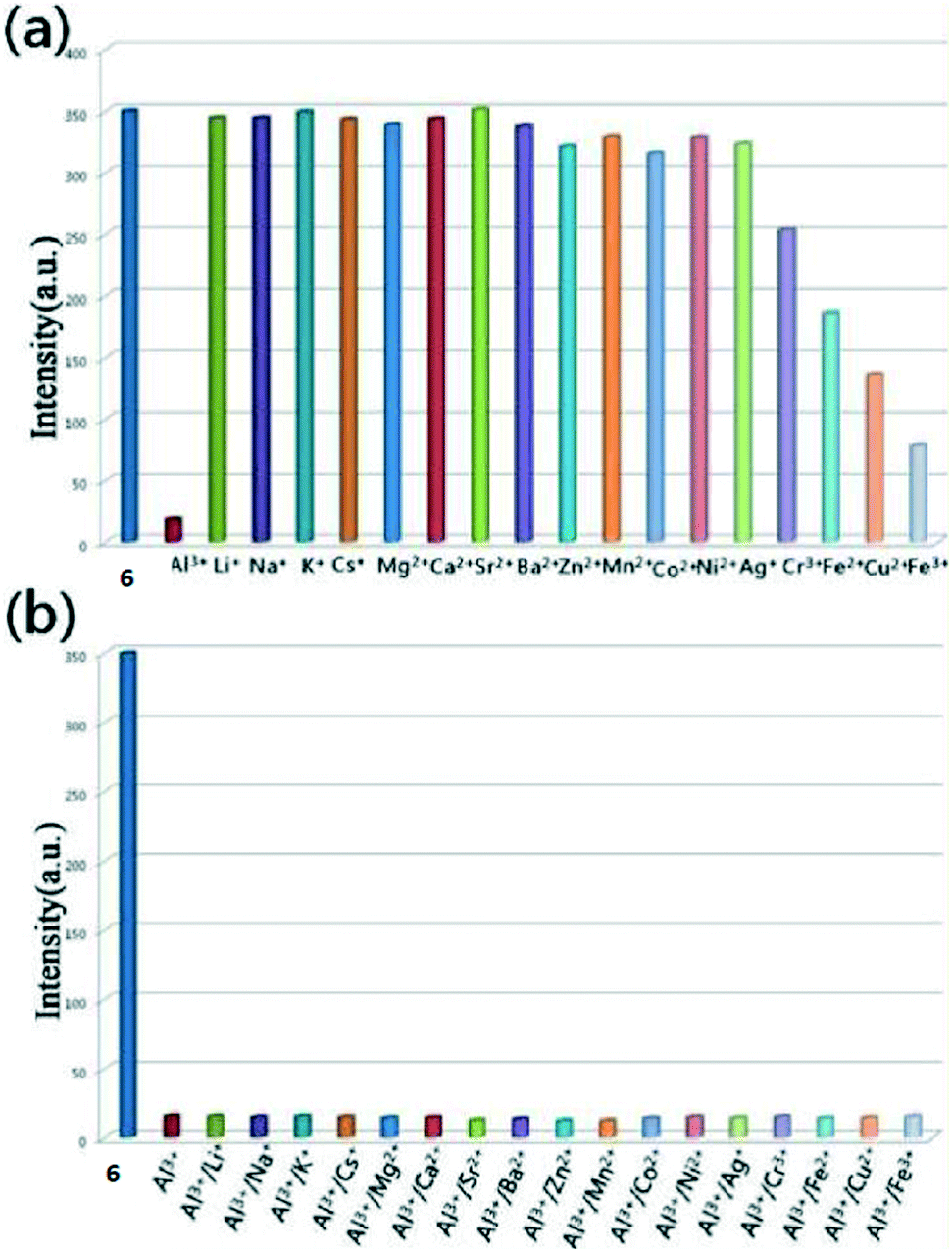 | ||
| Fig. 4 (a) Luminescence intensity of the 5D0 → 7F2 transitions (614 nm) of 6 in response to various cations (7 × 10−4 M) for 5 min. (b) Comparison of the luminescence intensity of 6 exposed to mixed cations (7 × 10−4 M). | ||
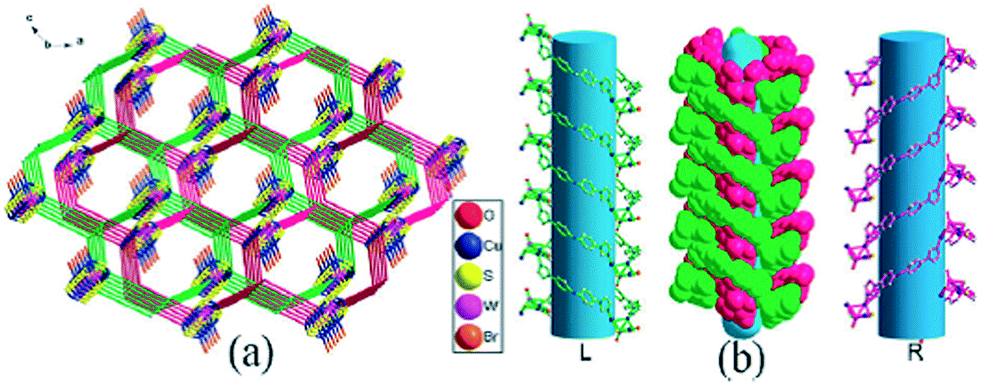
In 2015, one Cu(I) coordination network, namely, {[Cu6(bpz)6(CH3CN)3(CN)3Br]·2OH·14CH3CN}n (9), (bpz = 3,3′,5,5′-tetramethyl-4,4′-bipyrazole), was prepared by using a solvothermal method.13 The cyanide ligand in the network was generated in situ by cleavage of the C–C bond of CH3CN under solvothermal conditions. Compound 9 was a three-dimensional framework composed of a novel planar [Cu3Br] triangle and single Cu nodes, which were extended by μ2-bpz and μ2-CN to form a novel (3,9)-connected gfy network. Density functional theory calculations showed that single-electron delocalization of the Br atom induced the plane structure of [Cu3Br]. Owing to the large solvent-accessible void and more Cu(I) active sites, the network 9 suggested notable catalytic activity on the Click reaction, and 99% yield was achieved within 2 h.
Cluster–organic frameworks with tetranuclear secondary building units
M. Cindrić reported three isomorphous compounds based on cubane-like tetranuclear Ni(II) clusters14 differing in the type and amount of incorporated solvent and gave a detailed discussion about the correlation between structural, physical and chemical properties. Two starting compounds, {[Ni4L4(CH3OH)4]·0.32CH3OH·0.32H2O}n (10) and {[Ni4L4(CH3OH)4]·0.63CH3OH}n (11), were synthesized and used to generate a range of Ni4L4 clusters (H2L = tridentate Schiff base N-(2-hydroxy-5-methylphenyl)salicylideneimine). Conversions of coordinated and non-coordinated solvents and their effect on the magnetic and structural properties were examined. Loss of the coordinated solvent molecules caused the colour change to be red and yielded cluster 12, {[Ni4L4]}n (Scheme 2). Several cycles of annealing-MeOH sorption experiments showed that the process was not fully reversible. Namely, the geometry of the Ni4L4 core was shown to vary, and materials obtained by sorption processes were also found to differ, probably in the amount of non-coordinated solvent. Similarly, the powder diffraction pattern results suggested that compounds 10 and 11 showed differences when they were consecutively exposed to H2O and CH3OH vapors. This indicated that the number and amount of coordinated and non-coordinated solvents can vary and were not easy to predict. Moreover, according to quantum-mechanical calculations, it was the disorder that played an important role in determining the magnetic properties of the material. The existence of mixed solvates and the statistical and dynamic disorder of solvent molecules presented here indicated that there were multiple risks in the design of materials with desired magnetic properties. Detailed interpretation of the structural and magnetic correlations remains a challenge for future studies of these systems.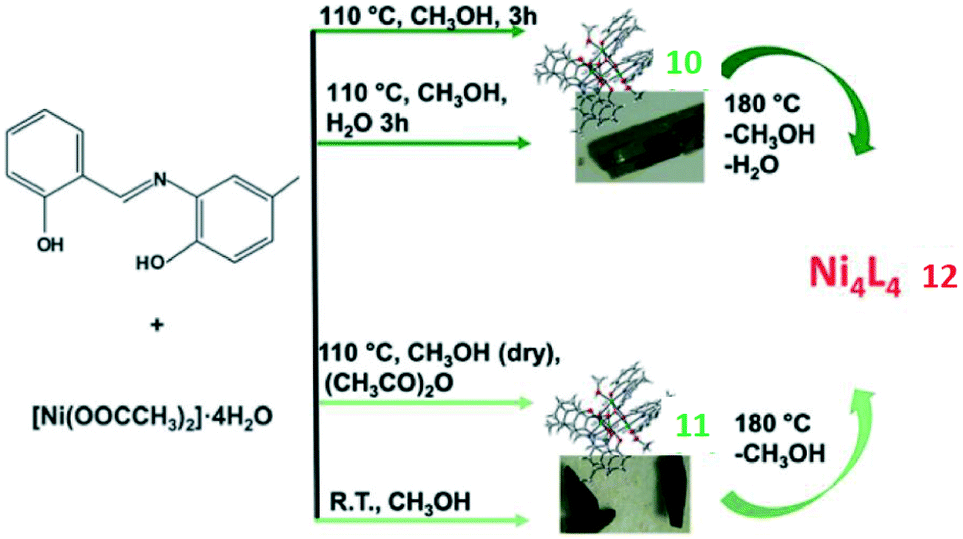 | ||
| Scheme 2 Synthesis of two green clusters {[Ni4L4(CH3OH)4]·0.32CH3OH·0.32H2O}n (10) and {[Ni4L4(CH3OH)4]·0.63CH3OH}n (11) and one red cluster of the formula {[Ni4L4]}n (12). | ||
Another example was reported by Y. Xiao about rare tetranuclear Zn clusters embedded into 3D energetic metal–organic framework channels by an enhanced extended hook method.15 The obtained metal–organic framework exhibited not only a high enthalpy of combustion but also strong green fluorescence. They innovatively combined nitrogen-rich energetic compounds (azide) into a new 3D MOF {[Zn4(N3)0.8(OH)1.2(BTEC)(HTZPC)2(H2O)2]·3.25H2O}n (13) (H2TZPC = 1-(2-(1H-tetrazol-5-yl)ethyl)piperidine-4-carboxylic acid, H4BTEC = 1,2,4,5-benzenetetracarboxylic acid) and implemented the encapsulation of rare tetranuclear Zn clusters [Zn4(μ1,1-N3)1.6(μ2-OH)2.4(CO2)4(H2O)4] into the framework channels of 13 by an extended tetrazolate–carboxylate hook ligand (Fig. 5). Notably, compound 13 exhibited a high enthalpy of combustion, low sensitivity, and strong green fluorescence.
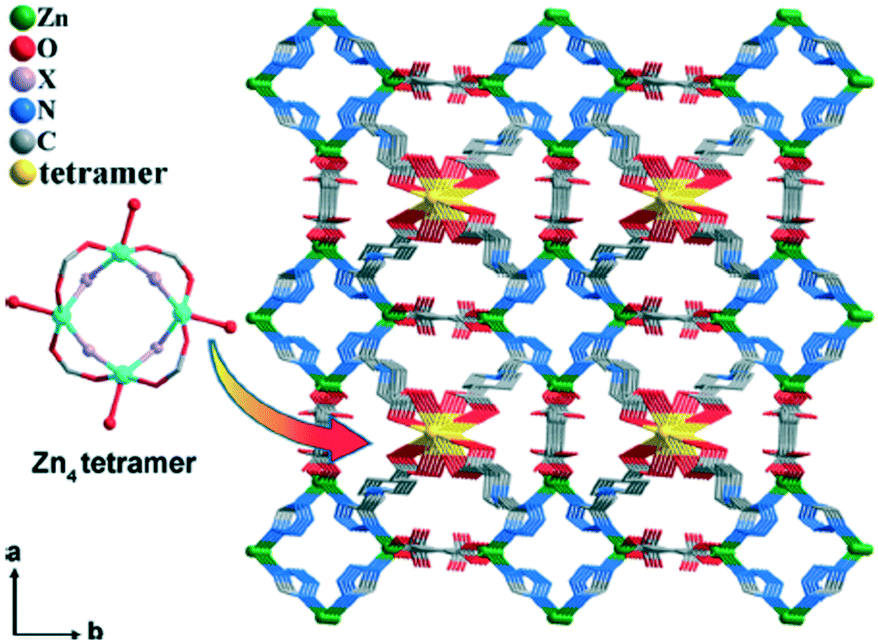 | ||
| Fig. 5 View of the 3D structure of 13 along the c axis. The Zn4 tetramer represents [Zn4(μ1,1-N3)1.6(μ2-OH)2.4(CO2)4(H2O)4]. | ||
Cluster–organic frameworks with pentanuclear or more nuclear secondary building units
In 2012, compound 14 with a pentanuclear secondary building unit, {[Co5O2(L)2(OBA)3]·7DMF}n (L = 1,1′-oxybis[3,5-dipyridine-benzene], H2OBA = 4,4′-oxybis(benzoic acid)), was synthesized by solvothermal reactions.16 Single-crystal X-ray analysis revealed that compound 14 crystallized in the space group C2/c. There were three independent octahedral Co2+ cations in the asymmetric unit: Co2 and Co3 were located at general positions, whereas Co1 lay on an inversion centre and one of the OBA ligands had the central oxygen O3 on a twofold axis. Co1 was coordinated to six oxygen atoms from four OBA anions and two μ3-O anions; Co2 was surrounded by three carboxylate oxygen atoms from three OBA anions and one μ3-O anion with two additional coordinations to L ligands; and Co3 was also coordinated to three carboxylate oxygen atoms and one μ3-O anion, leaving another two coordination sites occupied by N atoms from two L ligands. There were two μ3-O anions, which connected five Co cations to form a [Co5O2] cluster. The carboxylate groups from OBA anions further edged the [Co5O2] unit in the bi- and tridentate bridging modes to form a 4-connected 3D cds architecture, while, not considering the OBA, the L ligands connected the clusters to form a 4,8-connected 3D alb framework. Therefore, we can define the [Co5O2] cluster as a 12-connected node and the L ligand as a 4-connected node (Fig. 6). The 3D framework occupied only 56.9% of the total crystal volume, and the remaining space was occupied by the DMF molecules. To evaluate the permanent porosity of the framework, nitrogen sorption isotherm measurements were performed on the desolvated crystalline samples at 77 K. It exhibited type I sorption isotherms and takes up 175.4 cm3 g−1 of N2. Low-pressure H2 uptake was further measured at 77 K. Its uptake reached about 151.7 (1.35 wt%) at 850 mmHg, which was higher than that of MOF-177 (1.24 wt%).17 The result showed that the compound with permanent porosity exhibited a good affinity for hydrogen. Meanwhile, CO2 and CH4 isotherms at 273 K reached the maximum of 101.6 cm3 g−1 and 35.1 cm3 g−1, respectively. When the pressure was increased, the CO2 adsorption capacity greatly exceeded that of CH4. It was suggested that it had a potential application in gas separation processes of CH4/CO2. The median pore width was 7.142 Å; the selectivity for CO2/CH4 cannot be attributed to the pores restricting the adsorption of gases. The lower CH4 adsorption was due to its smaller quadrupole moment and lower polarisability than that of CO2.18 | ||
| Fig. 6 Schematic representation of 4,12-connected topology of compound 14. | ||
A highly connected 3D metal–organic framework 15 with tfz-d topology based on a hexanuclear Zn6O2 cluster and a flexible carboxylate ligand (H3L = 3,5-bis(4-carboxyphenoxy)benzoic acid) has been synthesized.19 It was formulated as {[Zn5(μ3-OH)4L2]·7H2O·DMF}n based on IR, TG, elemental analysis and an X-ray study. Single crystal X-ray diffraction study indicated that 15 had a 3D structure constructed from a Zn6O2 cluster and an L3− linker. The Zn6O2 cluster consisted of two Zn3O, and the core of the cluster consisted of a single O (μ3-OH) atom bonded to three Zn atoms, forming a regular Zn3O tetrahedral structure. The structure can be considered to be a two nodal (3,8)-connected 3D net with tfz-d topology type. Unlike the Zn4O node which is sensitive to H2O, the Zn6O2 can be resistant to water, hexane, acetonitrile, ethanol, DMF and toluene molecules at room temperature for a long time. The N2 isotherm at 77 K revealed that 15 exhibited typical type I adsorption behavior (indicative of homogeneous micropores), and adsorbs 89.6 cm3 g−1, corresponding to a Brunauer–Emmett–Teller (BET) surface area of 277.9 m2 g−1 and a Langmuir surface area of 367.9m2 g−1. H2 sorption measurements were also carried out; at 77 K and 850 mbar, the hydrogen uptake capacity of 15 was 4.48 mmol g−1. In addition, it took up different amounts of CO2 (56.8 cm3 g−1) and CH4 (26.2 cm3 g−1) at 273 K and 800 mbar, owing to the significant differences between gas molecule diameters (CO2 3.30 Å and CH4 3.758 Å) and the molecular sieving effect of the narrow apertures. The uptake of CO2 increased quickly at low pressure and reached 1.72 mmol g−1 at 100 mbar, while CH4 adsorption uptake was 0.51 mmol g−1, indicating the high selectivity of gas sorption for CO2 over CH4 under relatively low pressure. With further increase of the pressure, the sorption uptake reached 2.53 mmol g−1, which was in accordance with the fact that the narrow apertures could induce a stronger interaction with the host framework.20 At this temperature, both CO2 and CH4 molecules had sufficient kinetic energy to diffuse freely within the cages. At the same time, no hysteresis was observed for both gases, which might lead to applications in gas separation. The luminescence results suggested that the dynamic nature of 15 could allow for specific guest–host interactions with this linker, which may lead to potential applications in combined separation and detection experiments. Shifts in emission were observed upon changing the solvent, with peak maxima decreasing in energy in the order DMF > toluene > hexane > acetonitrile > ethanol > water. This response was found to be fully reversible through multiple cycles of exchanges between solvents.
Another example of a hexanuclear [Zn6O] cluster has been reported in 2012. Tris-(p-carboxyphenyl)-methane (H3TCOPM) was selected as a ligand to synthesize a new non-interpenetrating porous MOF with the formula {[Zn12(μ6-O)2(TCOPM)4]·3H2O·8NO3·8DMF}n (16⊃DMF).21 The core of the cluster consisted of a single O (μ6-O) atom bonded to six Zn atoms, forming a regular [Zn6(μ6-O)] polyhedron. A Zn6(μ6-O) cluster acting as a six-connected SBU linked by TCOPM3− acted as a triangular organic building block to generate the 3D framework. To our surprise, 16⊃DMF emitted a bright salmon pink fluorescence and can reversibly sense for small molecules. The robustness and the permanent micropore feature of the activated 16⊃DMF were established by a N2 sorption isotherm at 77 K with a BET surface area and pore volume of 533.8 m2 g−1 and 0.2389 cm3 g−1, respectively.
For 16⊃DMF, the most interesting feature was that its photoluminescence (PL) intensity was largely dependent on the solvent molecules, particularly in the case of DMF and THF, which exhibited the most significant enhancing and quenching effects, respectively. The intensity of the PL in 16⊃DMF depended on the identity of the guest molecule, with DMF > toluene > acetone > CHCl3 > propanol > acetonitrile > ethanol > methanol > 1,4-dioxane > THF (Fig. 7), which was probably due to different interactions with solvents from each other. Molecules with -OH groups were also effective quenchers for 16⊃DMF and the quenching effects of the PL increased with the decrease of the C chain. In addition, 16⊃DMF also responded to those solvent vapors, but at a much slower speed with their volatilization, for example, THF, methanol, DMF and so on.
 | ||
| Fig. 7 The PL spectra of 16 introduced to various pure solvents when excited at 390 nm and the photo of the PL: toluene, CHCl3, methanol, THF from left to right under UV light at 365 nm. (The inset shows the PL spectra of 16 in DMF, toluene, CHCl3, methanol, and THF solvents). | ||
In order to determine the fluorescence intensity response of 16⊃DMF in different solvents, PXRD tests were performed. After 16⊃DMF was soaked in toluene, 16⊃toluene was isolated. The structure of 16⊃toluene was similar to that of 16⊃DMF except that the a, b and c values of the unit cell were elongated from 22.5150(9) to 22.5597(12) Å. In the PXRD pattern of 16⊃toluene, the peak for the [200] reflection shifted to a lower 2θ value, corresponding to the slight shrinkage of d200. The maximum peak shift of 0.33 was observed in THF solvent. The breathing phenomenon of 16⊃DMF can provide an explanation for the intensity difference of PL between DMF and other solvents because of slight changes in structure. Thus the magnitude of the fluorescence intensity depended on changes in structure in different solvents. To gain further insight into the role of DMF, the activated 16⊃DMF can be achieved by outgassing the sample at 220 °C overnight under a vacuum. The color of 16⊃DMF changed from pink to yellow, and the peak for the [200] reflection also shifted to a lower 2θ value. For the activated 16⊃DMF, the emission intensity sharply decreased (at 579 nm and 613 nm) and finally became very small, which demonstrated that the solvent plays a key role in the PL in this region. It was interesting that, through the grinding method, the 579 and 613 nm emission bands disappeared for 16⊃DMF, which may be attributed to the loss of solvents.
Recently, Eddaoudi and workers demonstrated the successful use of reticular chemistry as an appropriate strategy for the design and deliberate construction of a zirconium-based metal–organic framework2217 with an intricate pbz underlying net topology (Fig. 8). The judicious selection of the perquisite hexagonal building units, six connected organic and inorganic building blocks, allowed the formation of compound 17, the first example of a Zr(IV)-based MOF with pbz topology. Prominently, pbz-MOF-17 was highly porous, with associated pore size and pore volume of 13 Å and 0.99 cm3 g−1, respectively, and offered high gravimetric and volumetric methane storage capacities (0.23 g g−1 and 210.4 cm3 (STP) cm−3 at 80 bar). Notably, the pbz-MOF-17 pore system permitted the attainment of one of the highest CH4 adsorbed phase density enhancements at high pressures (0.15 and 0.21 g cm−3 at 35 and 65 bar, respectively) as compared to benchmark microporous MOFs.
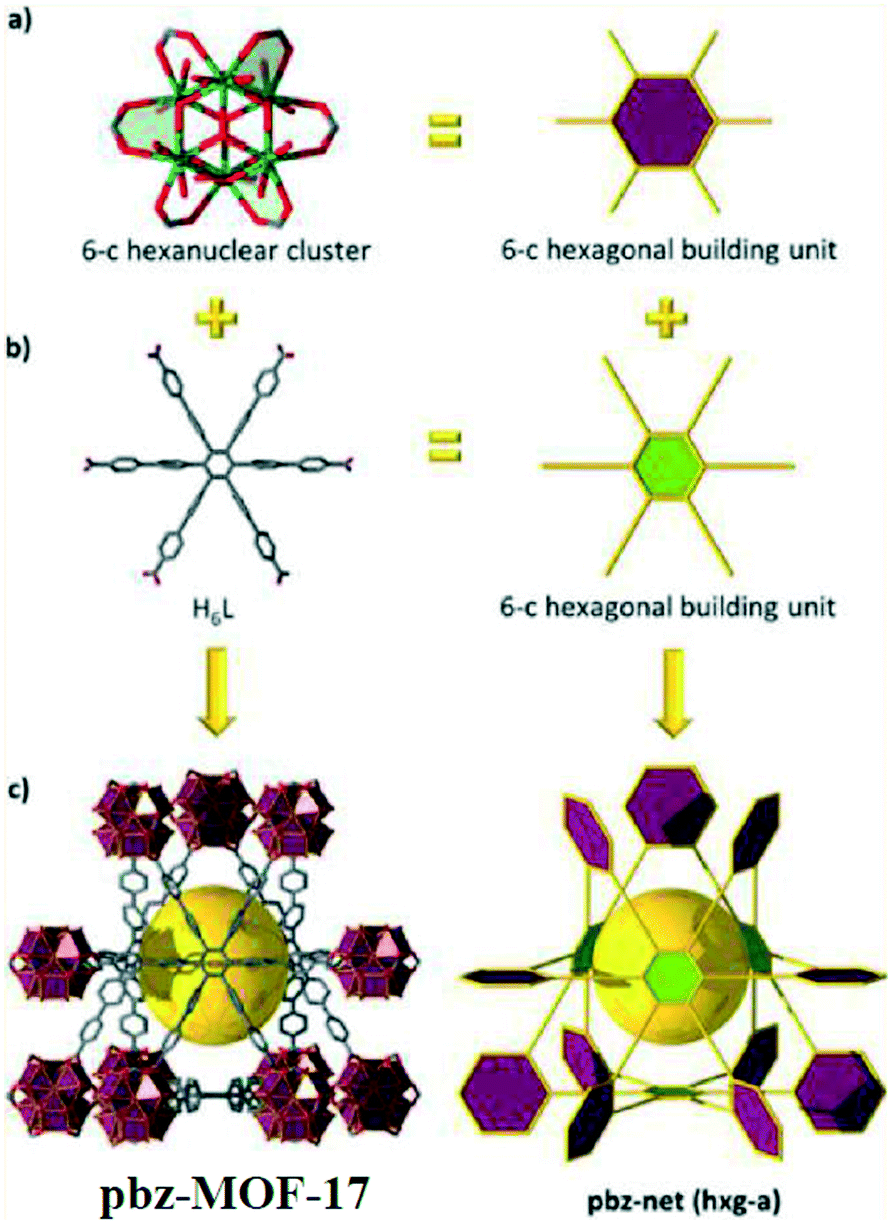 | ||
| Fig. 8 Combination of Zr6 clusters (a) with the hexatopic H6L ligand (b), both with hexagonal planar geometry, leads to the construction of pbz-MOF-17 (c), displaying the augmented hxg-a net, originally described in the structure of pbz. | ||
Cluster–organic frameworks with mixed types of secondary building units
A rare chiral 3D cluster–organic framework, {[Zn17O5(NTB)6(NDB)3]·41H2O}n (18) ((H3NTB = 4,4′,4′′-nitrilotrisbenzoic acid and H2NDB = 4,4′-nitrilodibenzoic acid)), was prepared and structurally characterized.23 The complex contained two types of [Zn4(μ4-O)(COO)6] and unreported [Zn9(μ3-O)3(COO)12] SBUs, which linked bi- and tricarboxylic acids and extended to an unprecedented (3,6,12)-connected framework. [Zn9(μ3-O)3(COO)12] as a unique cluster aroused our great interest. In order to scrutinize the nature of SBUs, electronic structure calculations were performed for model clusters Zn4H6(μ4-O)(COO)6 and Zn9H12(μ3-O)3(COO)12. The result indicated that the [Zn9(μ3-O)3(COO)12] configurations were greatly stabilized by linkers or dynamically stable in the MOFs. In such a special configuration, a smaller HOMO–LUMO energy gap (Fig. 9) will favorably influence the optical properties of the MOFs. | ||
| Fig. 9 Calculated energy diagram for clusters Zn4H6(μ4-O)(COO)6, Zn9H12(μ3-O)3(COO)12, and Zn9H12(μ3-O)(μ4-O)2(COO)12 by the B3LYP/ 6-311++G(d,p) (for clarity, hydrogen atoms were omitted). | ||
In 2011, we synthesized an unusual 3D structure with new topology based on Zn4O and Zn3–OH clusters, {[(Zn4O)1/2(Zn3–OH)1/2L3/2O1/4(H2O)5/2(NO2)1/2]·3(H2O)4}n (19) (H3L = 5′-carboxyl- [1,1′:3′,1′′-terphenyl]-4,4′′-dicarboxylic acid).24 The compounds with both independent Zn3–OH and Zn4O clusters have not been common, although many works reported MOFs with Zn3–OH or Zn4O clusters. While the coordination of water molecules, the Zn4O clusters were different from those reported, the μ4-oxo bridged Zn4O cluster was edge-bridged by five carboxylate groups from five L3− ligands and two coordinated water molecules and one O atom from nitrite anion to provide the Zn4O(L)5(H2O)2 unit, while the μ3-OH bridged Zn3O cluster was edge-bridged by four carboxylate groups from four L3− ligands and four coordinated water molecules and one O atom from nitrite anion to generate the (Zn3–OH)(L)4(H2O)4 (Fig. 10). There were three coordinated numbers of Zn in compound 19: Zn1 and Zn5 were five-coordinated, the τ trigonality factors were 0.098 and 0.329, respectively, indicating that they were in the distorted square pyramidal coordination geometry.25 Zn2 and Zn3 were tetrahedrally coordinated, Zn4 was octahedrally anchored by two carboxylate bridges, two coordinated water molecules, one O atom from nitrite anion and the central μ3-oxygen. The Zn4O clusters and Zn3–OH clusters were connected by nitrite anions, and two Zn4O clusters were connected by O bridging atoms; finally, they assembled L3− ligands to a 3D structure with 1D channels. The channels were occupied by solvent molecules. The desolvated framework showed 34.4% void space to the total crystal volume. To better understand the nature of this intricate framework, a topological approach has been applied; we reduced multidimensional structures to simple nodes and connection nets. (Zn4O)(L)5(H2O)2 and (Zn3-OH)(L)4(H2O)4 can be regarded as 7-connected and 5-connected nodes, respectively. All crystallographically independent L3− ligands acted as 3-connected linkers. Therefore, the whole structure can be represented as a new 3,3,3,5,7-c network topology.
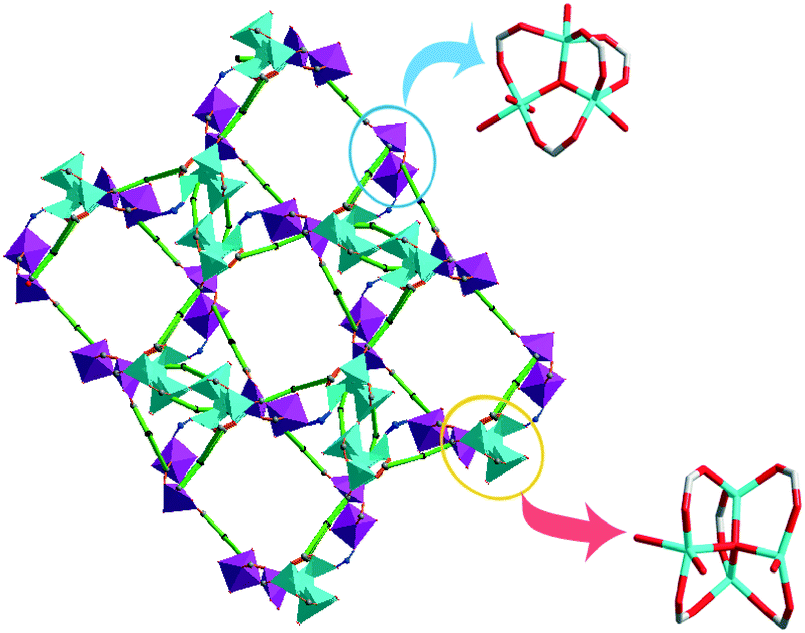 | ||
| Fig. 10 Zn3–OH and Zn4O clusters in compound 19 (Zn4O clusters are turquoise and Zn3O clusters are pink). | ||
Heterometallic coordination polymers
Heterometallic coordination polymers are now being increasingly investigated for opportunities to incorporate unusual metal coordination environments to enhance catalytic, photoluminescence, or other properties. The incorporation of two or more transition metals in a coordination polymer is a current challenge for designed synthesis. However, it was not very straightforward to obtain the heterometallic coordination polymers because it was difficult to have control over the self-assembly process. Besides, it was also difficult to assign accurate metal ions in determining the crystal structure.Metal–oxygen-based heterometallic coordination polymers
The appropriate ligand design is very important and multidentate bridging ligands are widely used to create small clusters, such as oxido, hydroxido, azido, water, and halogeno groups.26 The versatile binding sites of appropriate polytopic ligands facilitate connecting of more metal centers, resulting in the formation of polynuclear clusters. Chakraborty et al. have already reported that some polytopic ligands with well-defined and appropriately separated coordination compartments can achieve formation of the desired polynuclear clusters.27 Recently, for the first time, their team also successfully synthesized and reported a 3D single crystal of {[MnCu2(dpkO2H)2(dpkO2)N3]·(NO3)·H2O}n (20) (dpk = di-2-pyridyl ketone).28 In this 3D framework, the hexanuclear heterometallic Mn2(II)Cu4(II) clusters were connected through azido ligands, and the assembly of such Mn2(II)Cu4(II) clusters and ligands achieved a 3D framework (Fig. 11). The composition of CuII and MnII in the compound was confirmed from ICP-AES spectrometry and EDX spectrometry results. The SCXRD study revealed that the asymmetric unit contained two crystallographically different Cu(II) centers and one Mn(II) center. The positions of the metal atoms were assigned on the basis of the coordination geometry and structure refinement parameters. Magnetic studies were performed in detail and the compound showed a dominant antiferromagnetic interaction in the respective clusters, which was also supported by an extensive magneto-structural correlation which considered the different magnetic pathways.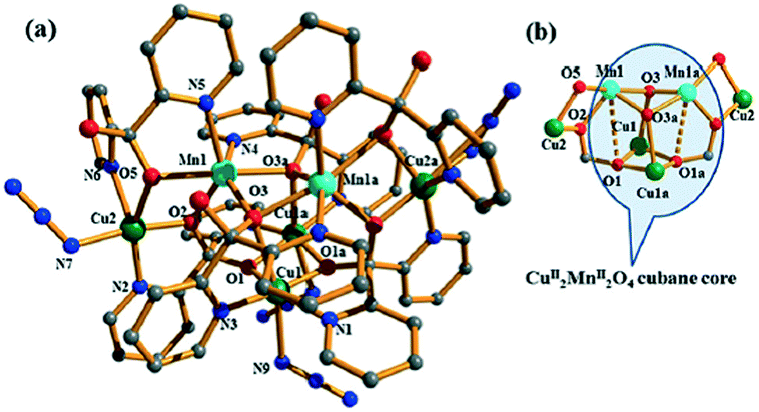 | ||
| Fig. 11 (a) The coordination environments around Cu(II) and Mn(II) centres. (b) The hexanuclear Mn(II)2Cu(II)4 core containing the cubane core. Color code: Cu: green; Mn: cyan; C: grey; O: red; N: blue. | ||
Another heterometallic coordination polymer was reported by Cheng et al.29 The solvothermal reaction of DyCl3·6H2O, Ni(NO3)2·6H2O, and H4abtc ligands (H4abtc = 3,3′,5,5′-azobenzene-tetracarboxylic acid), LiOH, and HNO3 in mixed DMF/H2O solvents (DMF = N,N-dimethylformamide) produced a three-dimensional Ni(II)–Dy(III) heterometallic coordination polymer formulated as {[NH2(CH3)2]2[NiDy2(HCOO)2(abtc)2]}n (21). The compound was an anionic framework, with the [NH2(CH3)2]+ counterions in the crystal lattice, which was in situ decomposed from DMF molecules under solvothermal conditions, as well as the HCOO− groups. The free program SHAPE 2.0 (ref. 30) analyses indicated that the most ideal coordination geometry of the nine-coordinated DyIII ion was muffin, which was conducive to determining the position of the metal ions. In 21, Dy(III) and Ni(II) ions interconnected through carboxylic O donors of abtc4− ligands to generate a linear trimer “hourglass”-type {NiDy2} cluster, and the adjacent trinuclear {NiDy2} units were bridged by HCOO− groups to give a 1D “ladder” chain, which was further bridged by abtc4− ligands to form a new topology with the point symbol of {32·42·52}{34·412·58·621}{45·6}. Alternating-current magnetic susceptibility results indicated that 21 exhibited frequency-dependent out-of-phase signals with two relaxation processes (Fig. 12), which suggested that it showed single-molecule magnet (SMM) behavior and represented the first example by using an SMM cluster as the building block to create a 3D Ni–Ln heterometallic coordination polymer, to the best of our knowledge.
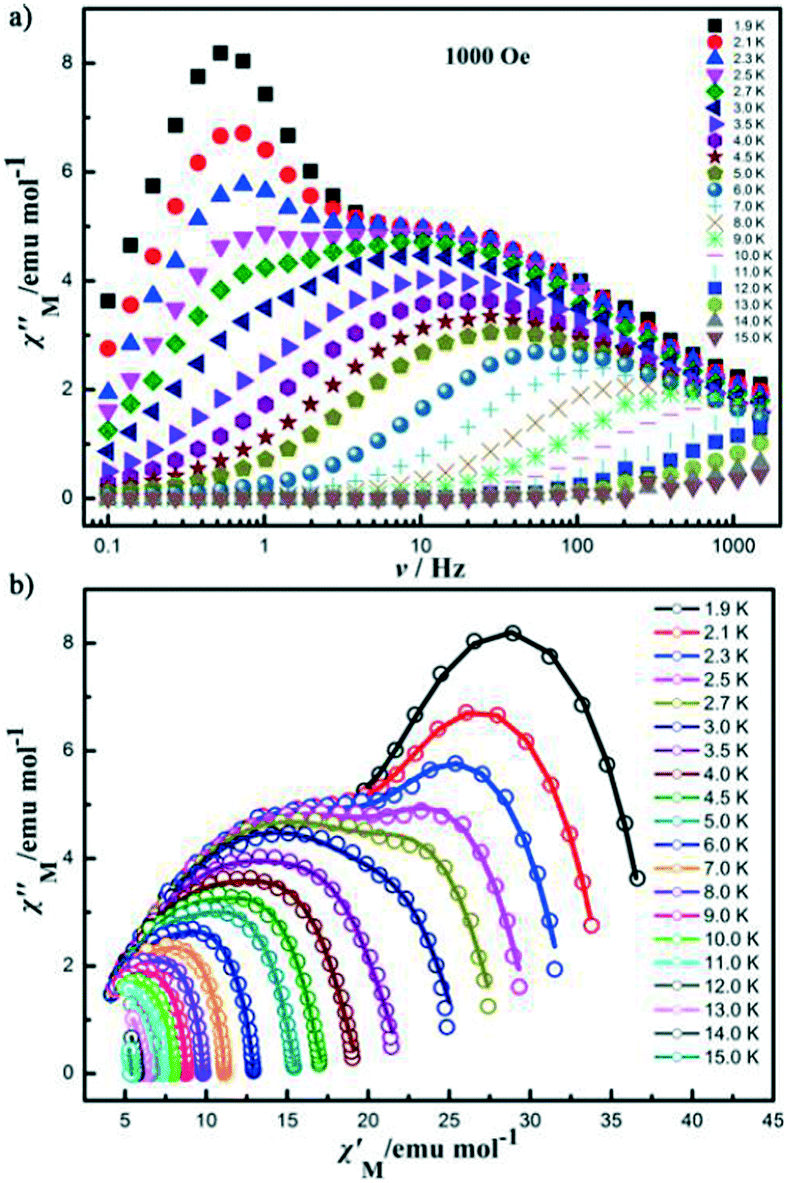 | ||
Fig. 12 (a) Temperature dependence of 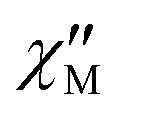 measured at different temperatures in a 1000 Oe dc field. (b) The Cole–Cole plots in the 1000 Oe dc field. The solid lines are the best fits. measured at different temperatures in a 1000 Oe dc field. (b) The Cole–Cole plots in the 1000 Oe dc field. The solid lines are the best fits. | ||
W–Cu–S-based heterometallic coordination polymers
In pursuit of novel MOFs, our group was attracted by a kind of heterothiometallic SBU of [MS4Cux](x−2)+ (M = W/Mo, x = 1–6), in which an MS42− dianion chelates Cu(I) atoms by the S atoms forming the basic structure of the SBU. In contrast to [Zn4O(COO)6] or [Cu2(COO)4] SBUs, the M/S/Cu SBU has a stable central core, MS42−; the number and/or arrangement of Cu(I) atoms around MS42− can be varied by changing the synthesis conditions and different combinations of MS42− and Cu+ afford SBUs with different geometry and connectivity. Moreover, the basic unit of such M/S/Cu SBUs can take different charges by combination of a certain number of anions, such as halide ions or CN−etc., forming cationic, anionic or neutral framework skeletons.In 2011, we used a long ligand, 4,4′-di(4-pyridyl)-biphenyl (dpbp), which was more than twice the length of 4,4′-bipy, to synthesize a novel 3D framework,31 {[WS4Cu4(dpbp)4]2+·[WS4Cu3(dpbp)2I2]−·I−·xSolvent}n (22), which was formed by the interpenetration of four anionic and four cationic frameworks. More interestingly, the anionic net was formed by the tetranuclear [WS4Cu3I2]− SBU, and the cationic net was formed by the pentanuclear [WS4Cu4]2+ SBU, forming an unprecedented 8-fold non-equivalent interpenetrated structure (Fig. 13).
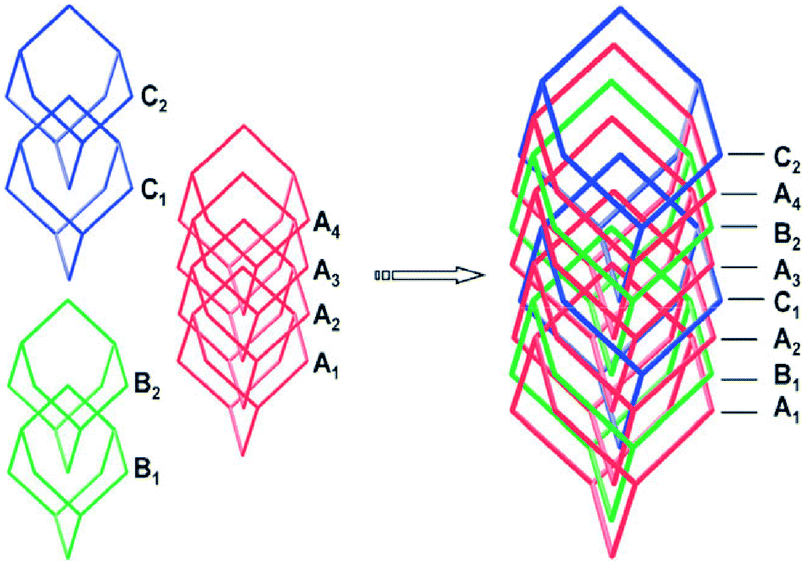 | ||
| Fig. 13 The assembly of the eight cationic (B and C) and anionic (A) frameworks being represented by the adamantane cages. | ||
Meanwhile, we reported two Mo(W)/S/Cu based cluster polymers {[MoOS3Cu3(bibp)2.5]·I}n (23) and {[WOS3Cu3Br(TIPA)]·H2O·DMF}n (24) (bibp = 4,4′-di(1H-imidazol-1-yl)-1,1′-biphenyl, tipa = tris(4-(1H-imidazol-1-yl)phenyl)amine).32 Compound 23 was a 2D cationic layer structure based on a twin-nest-shaped [Mo2O2S6Cu6]2− SBU which was composed of two nest-shaped clusters [MoOS3Cu3]+. Compound 24 was a 3D 2-fold interpenetrated framework which represented the first (10,3)-type MOFs based on Mo(W)/Cu/S SBUs (Fig. 14). The UV-vis absorption spectra of compounds 23 and 24 showed relatively low linear absorption in the visible and near-IR regions, suggesting that these compounds may have potential NLO properties.33 Thereby, we performed a Z-scan experiment to investigate their nonlinear optical properties. A reasonably good fit between the experimental data and the theoretical curves was achieved, and the nonlinear absorptive coefficients α2 were calculated to be 1.93 × 10−10 m W−1 for 23 and 2.81 × 10−10 m W−1 for 24, and the effective third-order nonlinear refractive indexes n2 of 23 and 24 were calculated to be 1.18 × 10−17 m2 W−1 and 1.38 × 10−17 m2 W−1, respectively. The results suggested that both 23 and 24 exhibited optical self-focusing behavior and reverse saturable absorption effects; they may therefore be promising candidates for third-order NLO materials.
 | ||
| Fig. 14 (a) Overall 2-fold interpenetrated (10,3)-b network of 24. (b) Schematic representation of each channel consisting of a left-handed helix and a right-handed helix. | ||
Another nanotubular metal–organic framework (MOF),34 {[(WS4Cu4)I2(dptz)3]·3DMF}n (25) (dptz = 3,6-di-(pyridin-4-yl)-1,2,4,5-tetrazine, DMF = N,N-dimethylformamide) for sensing small solvent molecules has been presented. In this structure, one WS42− dianion chelated four Cu+ atoms by the S atoms forming a saddle-shaped WS4Cu2+ heterothiometallic cluster unit. The WS4Cu2+ units acting as tetrahedral nodes were linked by two paired ligands and two single ligands with four adjacent units into a diamondoid network. When accommodating different solvent molecules as guests, the resulting inclusion compounds 25 exhibited different colors depending on the solvent guests, and more interestingly, the band gaps of these solvent-included complexes were in linear correlation with the polarity of the guest solvents. The solvent molecules can be sensed by the changes in the UV-vis spectra of the corresponding inclusion compounds, showing a new way of signal transduction as a new kind of sensor. The sensing by such a MOF appeared within the channel-containing material rather than on the external surface. When the dark red sample 25⊃DMF was immersed in CH3CN or CHCl3, the resulting compound 25⊃CH3CN changed to a bright red color, but 25⊃CHCl3 changed to totally black. Using other solvents, acetone, H2O, CH3OH, and C2H5OH, the resulting inclusion compounds changed to different colors depending on the solvent guests included (Fig. 15). While in solvent THF or DMSO, the porous compound 25 was slowly dissolved. The band gaps show excellent linear correlation with the ETN values of solvents. The best fits of band gaps against the ETN values were obtained by separately plotting nonhydroxylic solvents (slope, 2.989; correlation coefficient, 0.996) and hydroxylic solvents (slope, 0.607; correlation coefficient, 0.977). To estimate the effect of the dptz ligand on the solvatochromic response of 25, comparison tests were performed with an analogous MOF {[WS4Cu4(bpy)4][WS4Cu4(bpy)2I4]}n, (26, bpy = 4,4′-bipyridine), which was similar to 25 in both structure and composition.35 After the guest exchanging processes with the same solvents used for 25, the colors and UV-vis spectra of the obtained inclusion compounds 25⊃solvent revealed that no solvatochromic effect occurred for 26. The result indicated that the dptz ligand played an important role in the solvatochromic response of 25, which should be ascribed to its strong π-acceptor property and labile electronic structure to solvent polarity.
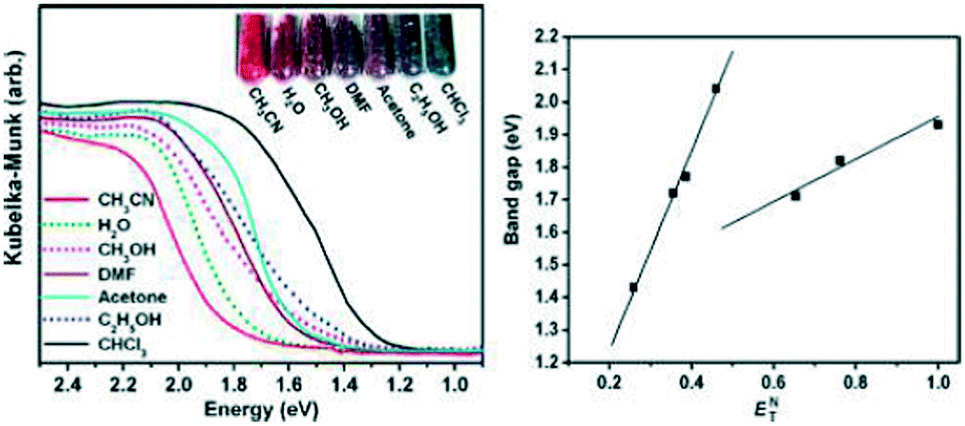 | ||
| Fig. 15 Left: the UV-vis spectra and photograph of the inclusion compounds 25⊃solvent. Right: band gaps of the inclusion compounds against the solvent ETN values. | ||
In 2012, four metal–organic frameworks (MOFs) formed by [WS4Cux]x−2 secondary building units (SBUs) and multi-pyridyl ligands were presented. The [WS4Cux]x−2 SBUs function as network vertexes showing various geometries and connectivities.36 Compound 27 contained one-dimensional channels formed in four-fold interpenetrating diamondoid networks with a hexanuclear [WS4Cu5]3+ unit as SBU, which showed square-pyramidal geometry and acts as a tetrahedral node. Compound 28 contained a brick-wall-like layer also with a hexanuclear [WS4Cu5]3+ unit as SBU. The [WS4Cu5]3+ unit in 28 was a new type of [WS4Cux]x−2 cluster unit in which the five Cu+ ions were in one plane with the W atom, forming a planar unit. Compound 29 contained large cages formed between two interpenetrated (10,3)-a networks also with a pentanuclear [WS4Cu4]2+ unit acting as a triangular node (Fig. 16). Compound 30 contained zigzag chains with a tetrahedral [WS4Cu3]+ unit as SBU, which acted as a V-shaped connector. The influence of synthesis conditions including temperature, ligand, anions of CuI salts, and the ratio of [NH4]2WS4 to CuI salt on the formation of these [WS4Cux]x−2-based MOFs has been studied. In most cases, the temperature and ligand were more important than the ratio of [NH4]2WS4 to CuI salt in synthesizing the [WS4Cux]x−2-based MOFs, and the effect of changing halide ions was minor if they just function as counterions or were involved in weak interactions, but important if they were involved in bridging interactions.
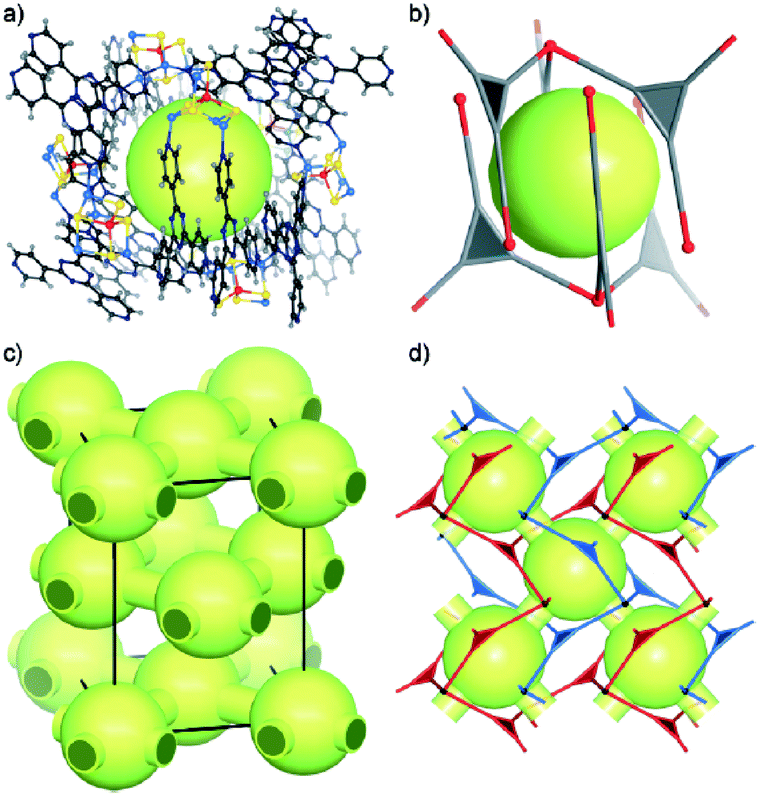 | ||
| Fig. 16 (a) The cage formed between two interpenetrated frameworks in 29. (b) Schematic representation of one cage. (c) The face-centered distribution of cages in the unit cell of 29. (d) Schematic presentation of the cages (large light green balls) and small channels (semi-transparent light green pipes) between them; the SBUs are depicted as small black balls and the tpt twin ligands as red or blue triangles. | ||
In 2013, three new heterothiometallic cluster nanoball compounds37 have been reported, {[(MS4Cu3)2(L)6](CuCl3)(H2O)8}n [M = W (30), Mo (31)] and {[(WS4Cu3)2(L)6](CuBr3)(H2O)7}n (32) (L = 1-(3-(1H-imidazol-1-yl)phenyl)-1H-imidazole), by the self-assembly reaction of the nest-shaped cluster block [MS4Cu3X3]2− with L as an angular ligand. The main difference among compounds 30, 31, and 32 existed in the anion portion. In compounds 30 and 31, the coplanar anion [CuCl3]2− was encapsulated in the center of the cationic nanosphere and stabilized via C–H⋯Cl hydrogen bonds with the distance of H⋯Cl ranging from 2.54 to 2.70 Å and the angle of C–H⋯Cl ranging from 146° to 177°. Each chloride anion of the [CuCl3]2− group acted as a 6-fold C–H⋯Cl hydrogen bond acceptor from two coordinated ligands of the host ball in compounds 30 and 31. However, [CuBr3]2− was situated outside the nanoball in compound 32, which was probably attributed to the following two reasons: firstly, [CuBr3]2− was larger than [CuCl3]2−; secondly, the electrostatic potential around the [CuCl3]2− anion was more negative than that of the [CuBr3]2− and cannot produce effective C–H⋯Br or C–H⋯S hydrogen bonding interactions. The W⋯W distances in compounds 30 and 32 were 12.3133(12) Å and 12.3962(18) Å, respectively, and the Mo⋯Mo distance in compound 31 was 12.3133(12) Å (Fig. 17). Although [CuCl3]2− was encapsulated in the nanoball, the distance of W⋯W in compound 30 was slightly shorter than that in compound 32. This may be attributed to the formation of many C–H⋯Cl hydrogen bonding and weak electrostatic interactions between the host [(WS4Cu3)2(L)6]2+ cation and the anion guest molecules.
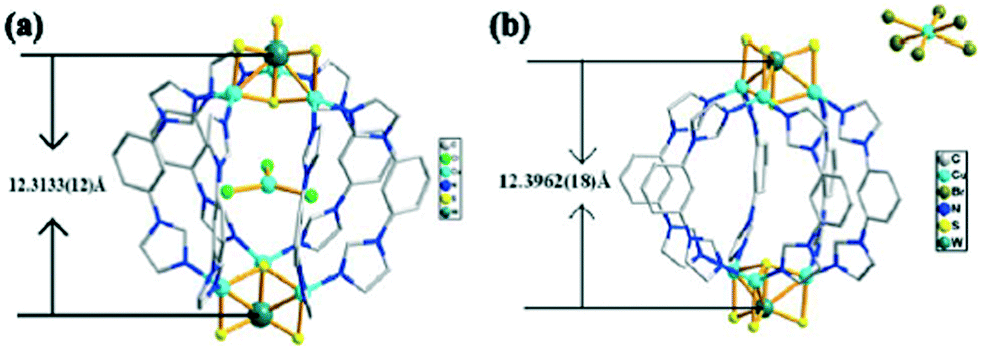 | ||
| Fig. 17 (a) Representation of the [CuCl3]2− guest encapsulated in the {[(WS4Cu3)2(L)6]}2+ cation nanocage in 30; (b) representation of the {[(WS4Cu3)2(L)6]}2+ cation nanocage in 32 with [CuBr3]2− sitting outside (solvent water molecules were all omitted for clarity). | ||
All compounds were insoluble in common organic solvents, and only slightly soluble in DMSO. The UV-vis absorption spectrum for compounds 30–32 showed that maximum absorption centered around 438 nm, 523 nm and 470 nm, respectively. The data were calculated from the reflectance data using the Kubelka–Munk function, α/S = (1 − R)2/2R, where R is the experimentally observed reflectance, the absorption coefficient, and S the scattering coefficient. According to transition type direct-gap semiconductors, the band gaps for compounds WCuCl and MoCuCl were about 2.99 and 3.045 eV, respectively. These results showed that these compounds may be classified as semiconductors.
It was interesting to find that micro- and nanoscale crystalline particles of 30 can be fabricated using solvent precipitation methods.38 The microrods of (30a) were prepared by dissolving 2 mg of 30 in 4 mL DMSO with an ultrasound for 60 min and injecting a high polarity solvent (chloroform). Scanning electron micrograph (SEM) images of (30a) revealed morphological microrods of 400–100 nm in length and 30–60 nm in diameter. Due to the anisotropic crystal structure of the hexagonal crystal system, the energetically favorable directions would preferentially grow to form the microrods. When the DMSO system was injected by a low polarity solvent (hexane), and other reaction conditions were kept constant, the SEM image exhibited mono-disperse olive-shaped particles (30b) with sizes of between 1.5 and 6.5 mm. It was found that a change in the environment of the polarity may induce a morphological change. It was reasonable to conclude that the morphology of the as-prepared particles can also be tuned by simply varying the polarity of the solvent.
The relationship between metal cluster units with the structures of coordination polymers
It was noteworthy that utilizing infinite rod-shaped or cluster secondary building units (SBUs) can reduce interpenetration, which provided MOFs with permanent porosity and rigid architectures.Three new Co-based MOFs with a nanosized tetradentate pyridine ligand (TPPBDA = N,N,N′,N′-tetrakis (4-(4-pyridine)-phenyl) biphenyl-4,4′-diamine),39 have been synthesized under hydrothermal conditions. For compound 33, a large cavity caused a 4-fold interpenetration of the network. Compound 34 revealed a non-interpenetrating three-dimensional framework based on the [Co2(μ2-H2O)(CO2)2] unit. Compound 35 was also a 2-fold interpenetrating 3D net based on the [Co2(CO2)2] units. The mononuclear or dinuclear cluster units were interconnected by TPPBDA and carboxylate co-ligands, resulting in interesting structural diversities and various degrees of interpenetration.
In 2015, three different Co-MOFs, [Co3(L)2(DPDP)]n (36), [Co(HL)(DPDP)]n (37) and {[Co(HL)(1/2DPDP)3(H2O)]·H2O}n (38) (H3L = 4,4′,4′′-(nitrilotris(methylene))tribenzoic acid, DPDP = 4,4′-(2,5-dibutoxy-1,4-phenylene) dipyridine) have been co-crystallized in a one-pot reaction.40 As shown in Fig. 18, compound 36 featured a non-interpenetrating 3D structure based on [Co3(CO2)6N2] clusters; compound 37 was a 2D network based on [Co2(CO2)2] clusters. Unlike 36 and 37, complex 38 was a mononuclear network, which was topologically classified as a uninodal 2-fold interpenetrating net with bnn topology. Through quantum chemical calculations of these systems, we endeavoured to optimize the synthesis conditions. Finally, by adjusting the ratio of the solvent, a single component crystal of the three compounds has been obtained.
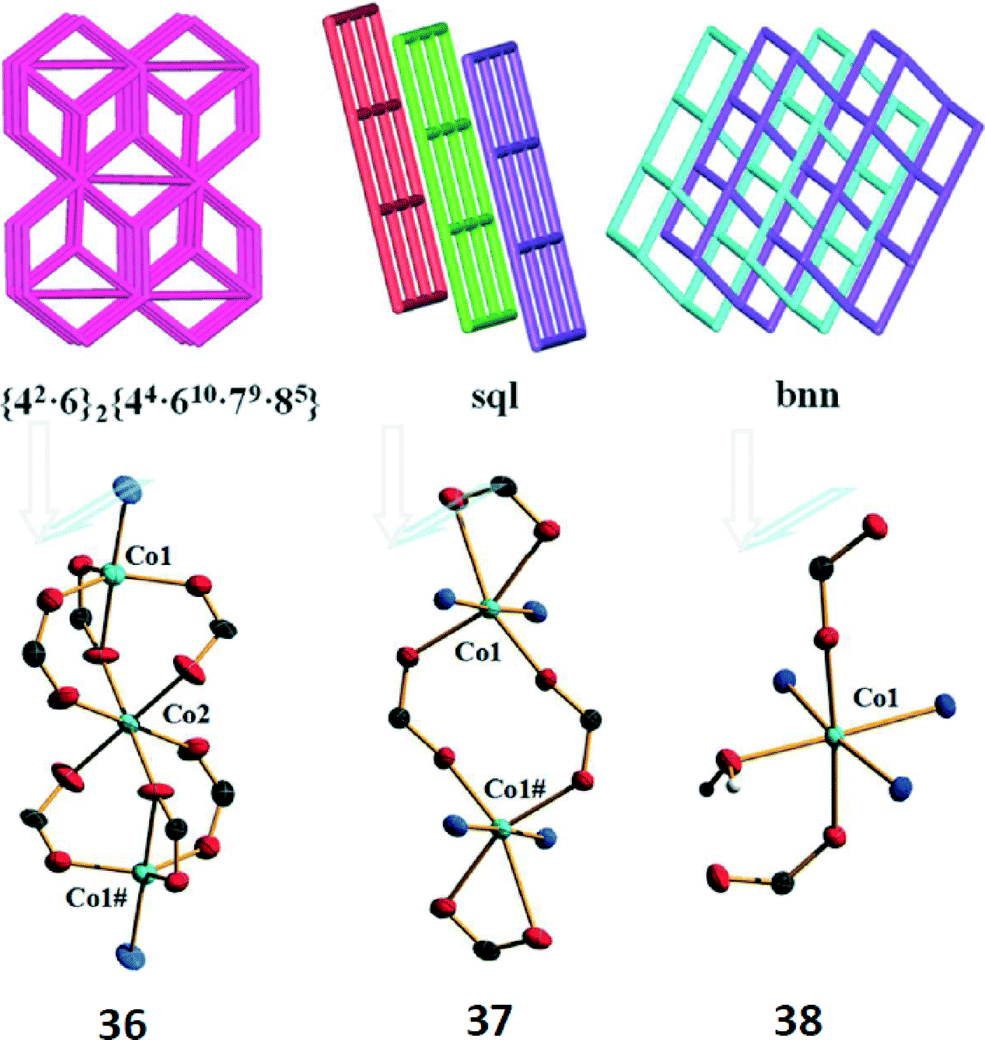 | ||
| Fig. 18 (Top) Topological representations of complexes 36–38. (Bottom) Different cobalt centers for complexes 36–38. | ||
Conclusions
The improved synthetic strategies of inorganic chemistry have led to numerous polynuclear clusters. More and more complicated structures will be synthesized and reported. In this article we have briefly highlighted some of the recent progress of homometallic and heterometallic MOFs and their diverse applications in sensing metal ions, small molecules and gases, nitroaromatic explosives, single-molecule magnet behavior, catalytic activity and nonlinear optical properties etc. In summary, metal cluster-based MOFs present themselves as a class of functional materials, which would continue to draw more interest and inquiry by both academia and industry. In the future, it is expected that the metal cluster-based MOFs would be applied in energy storage, batteries and supercapacitors, and the treatment of environmental pollution resulting from the development of industry.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 21601045, 21371092), National Basic Research Program of China (2010CB923303), China Postdoctoral Fund (2016M601768), and Fundamental Research Funds for the Central Universities (JZ2016HGTA0679).Notes and references
- K. S. Asha, R. Bhattacharjee and S. Mandal, Angew. Chem., Int. Ed., 2016, 55, 11528 CrossRef CAS PubMed
; J. He, N. W. Waggoner, S. G. Dunning, A. Steiner, V. M. Lynch and S. M. Humphrey, Angew. Chem., Int. Ed., 2016, 55, 12351 CrossRef PubMed
- H. He, J. Du, H. Su, Y. Yuan, Y. Song and F. Sun, CrystEngComm, 2015, 17, 1201 RSC
- S. C. McKellar, A. J. Graham, D. R. Allan, M. I. H. Mohideen, R. E. Morris and S. A. Moggach, Nanoscale, 2014, 6, 4163 RSC
- Q. X. Yang, X. Q. Chen, Z. J. Chen, Y. Hao, Y. Z. Li, Q. Y. Lu and H. G. Zheng, Chem. Commun., 2012, 48, 10016 RSC
- Q. X. Yang, X. Q. Chen, J. H. Cui, J. S. Hu, M. D. Zhang, L. Qin, G. F. Wang, Q. Y. Lu and H. G. Zheng, Cryst. Growth Des., 2012, 12, 4072 CAS
- A. Chanthapally, H. Yang, H. S. Quah, R. D. Webster, M. K. Schreyer, M. W. Wong and J. J. Vittal, Chem. – Eur. J., 2014, 20, 15702 CrossRef CAS PubMed
- L. Qin, M. X. Zheng, Z. J. Guo, H. G. Zheng and Y. Xu, Chem. Commun., 2015, 51, 2447 RSC
- L. Qin, Z. M. Ju, Z. J. Wang, F. D. Meng, H. G. Zheng and J. X. Chen, Cryst. Growth Des., 2014, 14, 2742 CAS
- Z. Q. Shi, Z. J. Guo and H. G. Zheng, Chem. Commun., 2015, 51, 8300 RSC
- Q. Zhang, A. Geng, H. Zhang, F. Hu, Z. H. Lu, D. Sun, X. Wei and C. Ma, Chem. – Eur. J., 2014, 20, 4885 CrossRef CAS PubMed
- S. S. Nagarkar, A. V. Desai and S. K. Ghosh, Chem. Commun., 2014, 50, 8915 RSC
- H. Xu, M. Fang, C. S. Cao, W. Z. Qiao and B. Zhao, Inorg. Chem., 2016, 55, 4790 CrossRef CAS PubMed
- Z. Xu, L. L. Han, G. L. Zhuang, J. Bai and D. Sun, Inorg. Chem., 2015, 54, 4737 CrossRef CAS PubMed
- M. Cindrić, G. Pavlović, D. Pajić, K. Zadro, D. Cinčić, T. Hrenar, E. Lekšić, A. B. P. Prieto, P. Lazićf and D. Š. Jung, New J. Chem., 2016, 40, 6604 RSC
- Y. Xiao, S. H. Wang, J. G. Xu, C. Sun, R. Li, Y. P. Zhao, Y. Yan, F. K. Zheng and G. C. Guo, CrystEngComm, 2016, 18, 5803 RSC
- L. Qin, J. S. Hu, M. D. Zhang, Z. J. Guo and H. G. Zheng, Chem. Commun., 2012, 48, 10757 RSC
- M. P. Suh, H. J. Park, T. K. Prasad and D. W. Lim, Chem. Rev., 2012, 112, 782 CrossRef CAS PubMed
- G. Ortiz, S. Brande's, Y. Rousselin and R. Guilard, Chem. – Eur. J., 2011, 17, 6689 CrossRef CAS PubMed
- J. H. Cui, Y. Z. Li, Z. J. Guo and H. G. Zheng, Chem. Commun., 2013, 49, 555 RSC
- L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294 RSC
- J. H. Cui, Z. Z. Lu, Y. Z. Li, Z. J. Guo and H. G. Zheng, Chem. Commun., 2012, 48, 7967 RSC
- D. Alezi, I. Spanopoulos, C. Tsangarakis, A. Shkurenko, K. Adil, Y. Belmabkhout, M. O'Keeffe, M. Eddaoudi and P. N. Trikalitis, J. Am. Chem. Soc., 2016, 138, 12767 CrossRef CAS PubMed
- J. S. Hu, L. Zhang, L. Qin, H. G. Zheng and X. B. Zhang, Chem. Commun., 2015, 51, 2899 RSC
- L. Qin, J. S. Hu, Y. Z. Li and H. G. Zheng, Cryst. Growth Des., 2011, 11, 3115 CAS
- A. A. W. Addison, T. N. Rao, J. Reedjik and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC
- C. D. Nicola, Y. Y. Karabach, A. M. Kirillov, M. Monari, L. Pandolfo, C. Pettinari and A. J. L. Pombeiro, Inorg. Chem., 2007, 46, 221 CrossRef CAS PubMed
-
(a) A. Chakraborty, K. L. Gurunatha, A. Muthulakshmi, S. Dutta, S. K. Pati and T. K. Maji, Dalton Trans., 2012, 41, 5879 RSC
- A. Chakraborty, A. Escuer, J. Ribas and T. K. Maji, Dalton Trans., 2016, 45, 15523 RSC
- S. Zhang, H. Li, E. Duan, Z. Han, L. Li, J. K. Tang, W. Shi and P. Cheng, Inorg. Chem., 2016, 55, 1202 CrossRef CAS PubMed
-
(a)
SHAPE, version 2.0, Electronic Structure Group, Universiat de Barcelona, Barcelona, Spain, 2010 Search PubMed
- Z. Z. Lu, R. Zhang, Y. Z. Li, Z. J. Guo and H. G. Zheng, Chem. Commun., 2011, 47, 2919 RSC
- X. Q. Yao, Z. R. Pan, J. S. Hu, Y. Z. Li, Z. J. Guo and H. G. Zheng, Chem. Commun., 2011, 47, 10049 RSC
- C. Zhang, Y. Cao, J. F. Zhang, S. M. T. Matsumoto, Y. L. Song, J. Ma, Z. X. Chen, K. Tatsumi and M. G. Humphrey, Adv. Mater., 2008, 20, 1870 CrossRef CAS
- Z. Z. Lu, R. Zhang, Y. Z. Li, Z. J. Guo and H. G. Zheng, J. Am. Chem. Soc., 2011, 133, 4172 CrossRef CAS PubMed
- K. Liang, H. G. Zheng, Y. L. Song, M. F. Lappert, Y. Z. Li, X. Q. Xin, Z. X. Huang, J. T. Chen and S. F. Lu, Angew. Chem., Int. Ed., 2004, 43, 5776 CrossRef CAS PubMed
- Z. Z. Lu, R. Zhang, Z. R. Pan, Y. Z. Li, Z. J. Guo and H. G. Zheng, Chem. – Eur. J., 2012, 18, 2812 CrossRef CAS PubMed
- L. Qin, Z. R. Pan, L. W. Qian, Y. Z. Li, Z. J. Guo and H. G. Zheng, CrystEngComm, 2013, 15, 5016 RSC
- G. Z. Yuan, C. F. Zhu, Y. Liu, Y. Fang and Y. Cui, Chem. Commun., 2010, 46, 2307 RSC
- M. X. Zheng, X. J. Gao, C. L. Zhang, L. Qin and H. G. Zheng, Dalton Trans., 2015, 44, 4751 RSC
- C. L. Zhang, Y. L. Li, T. Wang, Z. M. Ju, H. G. Zheng and J. Ma, Chem. Commun., 2015, 51, 8338 RSC
| This journal is © The Royal Society of Chemistry 2017 |