
TTF–PTM dyads: from switched molecular self assembly in solution to radical conductors in solid state
Manuel
Souto
,
Concepció
Rovira
,
Imma
Ratera
* and
Jaume
Veciana
 *
*
Institut de Ciència de Materials de Barcelona (ICMAB-CSIC)/CIBER-BBN, Campus de la UAB, 08193, Bellaterra, Spain. E-mail: iratera@icmab.es; vecianaj@icmab.es
First published on 14th September 2016
Abstract
Organic donor–acceptor (D–A) systems formed by the electron-donor tetrathiafulvalene (TTF) linked to the electron-acceptor perchlorotriphenylmethyl (PTM) radical through different π-conjugated bridges exhibit interesting physical properties such as bistability in solution or conductivity in solid state. Understanding the interplay between intra- and intermolecular charge transfer processes in solution is of high interest in order to rationalize the self-assembling ability and conducting properties of such dyads in solid state. In this Highlight we examine the self-assembling properties of different TTF–π–PTM radical dyads that have potential applications as molecular switches or conductors in the field of molecular electronics.
Introduction
During the last decades, the molecular approach to material science has emerged as a promising research field for the generation of new electronic devices at the nanoscale level because of their chemical and physical peculiarities.1 Owing to important perspectives in both fundamental sciences and applications in molecular electronics, numerous efforts have been made to design new multifunctional materials which involve the interplay between multiple physical properties (i.e. electrical, optical, and magnetic properties).2Multifunctional molecular materials can be even more interesting if they are able to switch between two or more states modifying their physical properties (i.e. optical, magnetic or electrical properties) upon the application of external stimuli (i.e. temperature,3 pressure,4 light5 or an electric field6). The interest of such phenomena is mainly due to their potential application as molecular switches and shape memory materials.7,8 Some molecular materials exhibiting bistability are, for example, spin-crossover,9 valence-tautomeric10 or donor–acceptor (D–A) systems.11
In this direction, there are some stable radicals that can exhibit reversible dimerization in response to external stimuli, with monomers and dimers coexisting at equilibrium in solution12 and may produce bistable systems in the solid state.13–15 Spirobisphenalenyl16,17 and thiazyl18,19 radicals are good examples of such materials (Fig. 1). These radicals are able to switch between a paramagnetic monomeric radical form at high temperatures and a diamagnetic dimeric form at lower temperatures; this mainly arises from the π–π intermolecular interactions within the dimers. These types of radical molecules are interesting not only because of the supramolecular bistability phenomenon in solution but also due to their potential use as single-component conductors once crystallized. Indeed, it was proposed that neutral organic radicals could be used as molecular conductors since the unpaired electron can serve as a charge carrier.20 However, most organic radicals exhibit a large Coulomb energy repulsion U and a narrow electronic bandwidth W giving rise to Mott insulator behavior. For this reason, most of the reported examples of neutral radical conductors are based on highly delocalized systems in order to decrease the repulsion energy U or incorporate heteroatoms to enhance the electronic bandwidth W.21–26
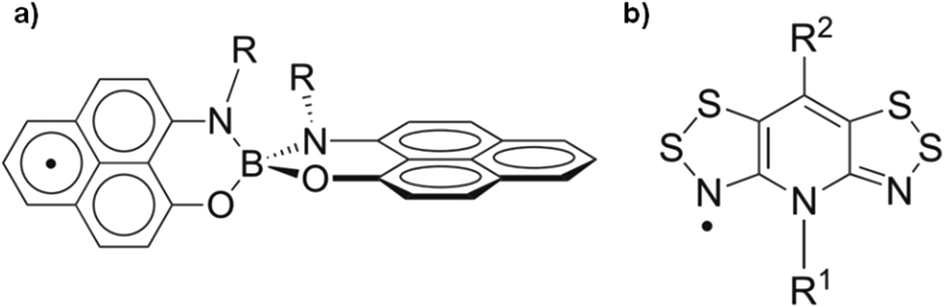 | ||
| Fig. 1 Molecular structures of a) the spirobisphenalenyl radical (R = n-Bu) and b) the bis(1,2,3-dithiazolyl) radical (R1 = Et, R2 = F). | ||
Organic donor–acceptor (D–A) systems
Organic molecules formed of electron donor (D) and electron acceptor (A) units covalently linked by a π-conjugated bridge (D–π–A) (Fig. 2) are an interesting family of molecules widely investigated for several applications, ranging from organic light-emitting devices,27 nonlinear optics,28 molecular electronics29 and photonics.30 D–A dyads are also very interesting materials due to the possibility of switching between different electronic states through variation of an external stimulus such as temperature, light or pressure. Indeed, when an appropriate external stimulus is applied, an intramolecular electron transfer (IET) process can be induced with the movement of an electron from the donor to the acceptor forming a charge-separated (zwitterionic) state that will show different physical properties (i.e. electrical, optical or magnetic properties). Thus, D–A systems could be potentially used as molecular switches due to the related bistability phenomenon.31 Otherwise, D–A dyads can also be of high interest if they exhibit molecular packing in the solid state with complete segregation of the donor and acceptor units exhibiting a good overlap between the molecules, as this is a prerequisite for obtaining organic conductors, semiconductors or organic field-effect transistors (OFETs) that could exhibit ambipolar characteristics.32–34 | ||
| Fig. 2 Schematic drawing of a D–A dyad and the associated bistability phenomenon when an external stimulus (i.e. T, P or light) is applied forming the zwitterionic state with different physical properties. | ||
Building blocks: TTF and the PTM radical
The electron-donor tetrathiafulvalene (TTF) and the electron-acceptor the perchlorotriphenylmethyl (PTM) radical are very good candidates as molecular building blocks for obtaining multifunctional D–A systems, since they allow the combination of magnetic, optical, conducting and electrochemical properties. Indeed, both units present different structural and electronic characteristics that make them interesting to build new multifunctional molecular materials.TTF unit
TTF and its various derivatives are among the most versatile molecules which exhibit interesting redox properties and remarkable electron donor character.35,36 TTF is a non-aromatic 14-π-electron system in which oxidation to the radical cation and dication species occurs sequentially and reversibly at very low oxidation potentials (E1/21 = 0.37 V and E1/22 = 0.67 V in CH2Cl2) (Fig. 3). These organosulfur molecules became of high interest due to their potential application in molecular electronics as semiconductors, superconductors and organic metals37 after the first synthesis of TTF reported by Wudl in 1970.38 Since then, several TTF derivatives have been reported to find applications in molecular and supramolecular structures, in switchable processes,39 as electron-donor units in D–A systems33,40 and for the preparation of unimolecular rectifiers on surfaces.41 Also, several studies on the dimerisation phenomenon in solution have been reported in which TTF is in the oxidized radical cation state.42–44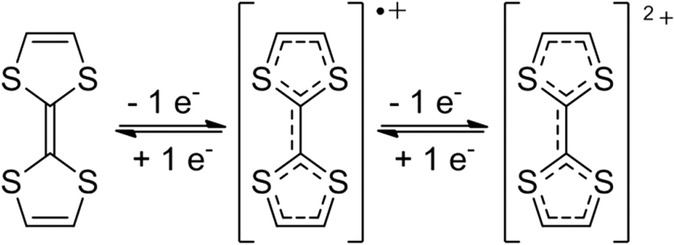 | ||
| Fig. 3 TTF and its radical cation and dication derivative species. | ||
PTM radical unit
PTM radicals are neutral open-shell organic molecules with a net magnetic moment which are characterized by an extremely high persistence and stability. They are composed of three chlorinated phenyl rings connected to a central carbon with sp2 hybridization exhibiting a large chemical and thermal stability. The large persistence of these radicals is mainly due to the presence of bulky chlorine atoms located in the ortho position providing a source of steric protection for the central carbon where the spin density is mainly localized.31,45 In addition, PTM radicals are also interesting because they are an electroactive species that can be oxidized to the perchlorotriphenylcarbonium ion or reduced to the PTM-carbanion (Fig. 4). The reduction of PTM radical occurs at very low potentials (−0.2 V in CH2Cl2) and it is more feasible than the corresponding oxidation that requires high potentials (1.6 V in CH2Cl2). Such evidence renders this type of radical optimal for the electron-acceptor unit in D–A systems to study IET to make switchable multifunctional molecular materials since the magnetic, optical and electronic properties of these open-shell molecules can be switched on and off with an external electrochemical stimulus.31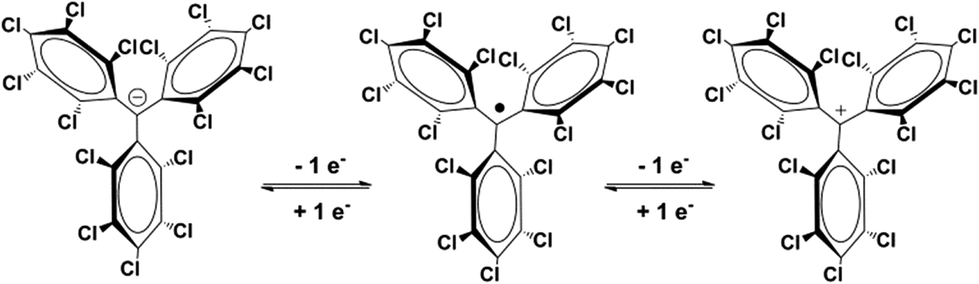 | ||
| Fig. 4 PTM radical and its anion and cation derivative species. | ||
In this Highlight we will review the recent developments in the synthesis and characterization of different TTF–π–PTM dyads (Fig. 5) which exhibit different physical properties such as optical, magnetic or electric properties depending on their molecular and supramolecular structure. In particular, we will focus on the study of the interplay between the intra- and intermolecular charge transfer (CT) processes which play a key role in the self-assembly of such dyads in solution in order to extend their physical properties and design molecular conductors in solid state.
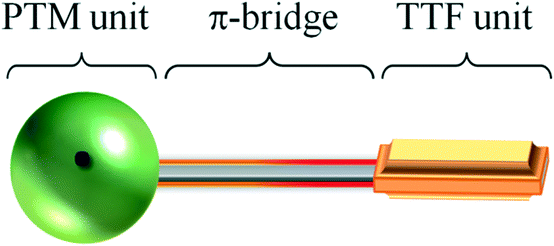 | ||
| Fig. 5 Schematic representation of a TTF–π–PTM dyad. | ||
Studies in solution
D–A radical dyads
Organic molecules that contain electron donor and electron acceptor units linked by π-conjugated bridges are very promising molecular materials for the development of molecular switches and memory devices.11 For example, during the last years in the framework of our group, it has been reported that different D–A systems based on the electron-acceptor PTM radical linked to ferrocene (Fc) derivatives can be switched from the neutral to the zwitterionic state by changing the polarity of the solvent or by tuning the temperature in the solid state (Fig. 6).46–51 The cooperativity of the electrostatic intermolecular interactions explains the bistability observed in such D–A molecular crystals.48,52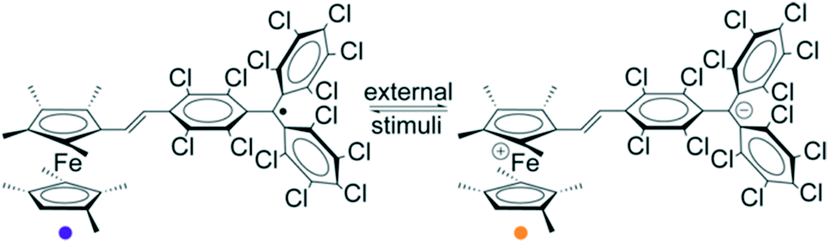 | ||
Fig. 6 Molecular structures relevant to the neutral D–A (purple,  ) and zwitterionic D+–A− (orange, ) and zwitterionic D+–A− (orange,  ) states of the Me8Fc-PTM derivative. Adapted with permission from ref. 50. Copyright (2013) American Chemical Society. ) states of the Me8Fc-PTM derivative. Adapted with permission from ref. 50. Copyright (2013) American Chemical Society. | ||
On the other hand, Nishida et al. proposed a TTF-based dyad which exhibits bistability in solution originating from an IET process between a TTF electron-donor linked to the electron-acceptor di-tert-butyl-6-oxophenalenoxyl (6OP) organic radical (Fig. 7).53 Such bistability was induced by changes in temperature and in the polarity of the solvent. Indeed, the phenomenon was followed by ultraviolet-visible-near infrared (UV-vis-NIR) spectroscopy observing the neutral state in CH2Cl2, whereas the zwitterionic state of the molecule was obtained in the polar CF3CH2OH solvent.
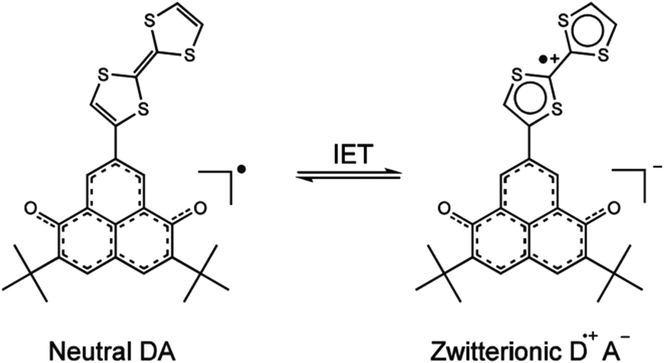 | ||
| Fig. 7 The two possible states (neutral and zwitterionic) of the TTF–6OP dyad. Adapted with permission from ref. 53; Copyright 2005, John Wiley and sons. | ||
Self-assembly of a TTF–PTM radical dyad (1)
In a similar approach to the previous examples, in our group we have reported a D–A dyad that consists of the electron-acceptor PTM radical linked to a TTF through a vinylene bridge (1).54 This organic D–A radical dyad exhibited a reversible switching from its neutral to its zwitterionic state that can occur through an IET process from the TTF to the PTM radical simply by modification of the solvent polarity (Fig. 8). For example, in the non-polar CH2Cl2 solvent only the neutral (1a) species was present, whereas in the polar dimethylformamide (DMF) solvent an IET process was promoted and the zwitterionic (1b) species was formed.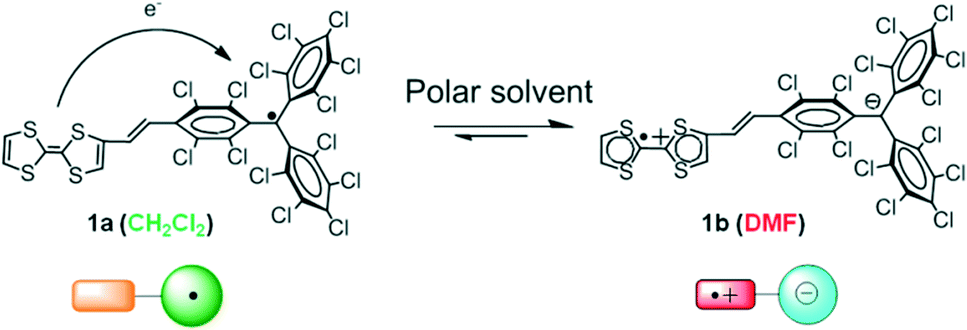 | ||
| Fig. 8 Schematic representation of the reversible switching of the radical dyad TTF–PTM 1 between the neutral (1a) and zwitterionic (1b) states through an IET process. | ||
This phenomenon was followed by UV-vis-NIR spectroscopy where the absorption spectrum of 1 in CH2Cl2 showed an intense band at 387 nm attributed to the radical chromophore of the PTM subunit, whereas the spectrum in DMF didn't show such a band but presented an intense band at 512 nm which corresponded to the anionic form of the PTM moiety (Fig. 9). Interestingly, the spectrum of 1 in acetone simultaneously exhibited a band located at 387 nm as well as a band at 505 nm, indicating the coexistence of both species 1a and 1b at equilibrium in this solvent.
 | ||
| Fig. 9 UV-vis-NIR spectra of dyad 1 in CH2Cl2 (green line), acetone (blue line), and DMF (red line). Inset: Low-energy range of the absorption spectra and colors of 1 exhibited in a) CH2Cl2, b) acetone, and c) DMF solutions. Adapted with permission from ref. 54; Copyright 2012, John Wiley and sons. | ||
In addition, it was demonstrated that the IET process promoted self-assembly of the dyads in the zwitterionic states to form diamagnetic dimers in DMF at room temperature. Indeed, the electron spin resonance (ESR) spectrum of dyad 1 in CH2Cl2 presented the typical signal for the PTM radical in agreement with the presence of the neutral species, whereas the spectrum in DMF didn't give any signal (Fig. 10). The lack of signal relating to the TTF+˙ cation radical suggested that the zwitterionic species aggregate forming diamagnetic dimers (π-dimer) that are ESR silent at room temperature.
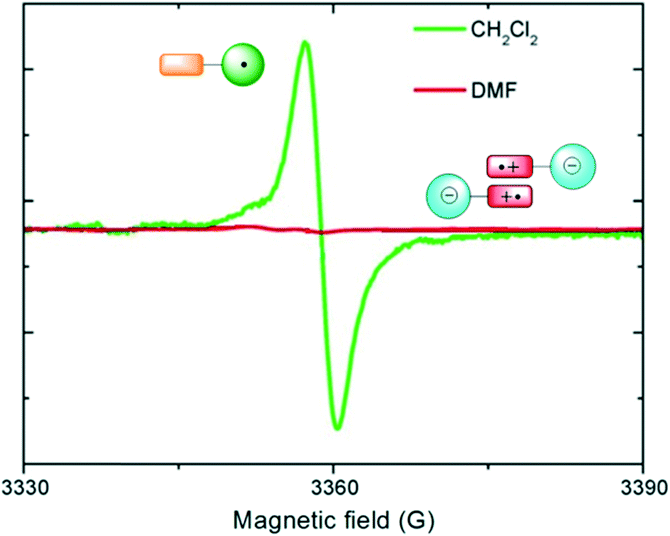 | ||
| Fig. 10 ESR spectra of dyad 1 at 300 K in CH2Cl2 (green line) and DMF (red line). Schematic representation of monomers 1a in CH2Cl2 and diamagnetic π-dimers of 1b in DMF solutions. | ||
Thus, dyad 1 exhibits an IET phenomenon that enables the reversible switching from its neutral state 1a to its zwitterionic state 1b simply by modification of the solvent. In addition, the TTF-based self-assembly of this dyad was induced by the IET process between the donor and acceptor groups, forming diamagnetic dimers of 1b in DMF solution.
Moreover, in order to investigate the origin of such an aggregation phenomenon, temperature-dependent optical and ESR spectroscopies studies in CH2Cl2 were performed for a family of TTF–PTM dyads bearing different number of electrons (i.e. oxidizing the TTF subunit to TTF+˙ or reducing the PTM˙ radical subunit to a PTM− anion) and/or with a hydrogenated PTM residue (Fig. 11) giving information on the formation of homo- and mixed-valence dimers.55 Analysis of the equilibrium constants and thermodynamic parameters showed that dimers formed by radical dyads are much more stable due to the contribution of the IET that delocalizes the electrons along the vinylene bridge and the PTM subunit.
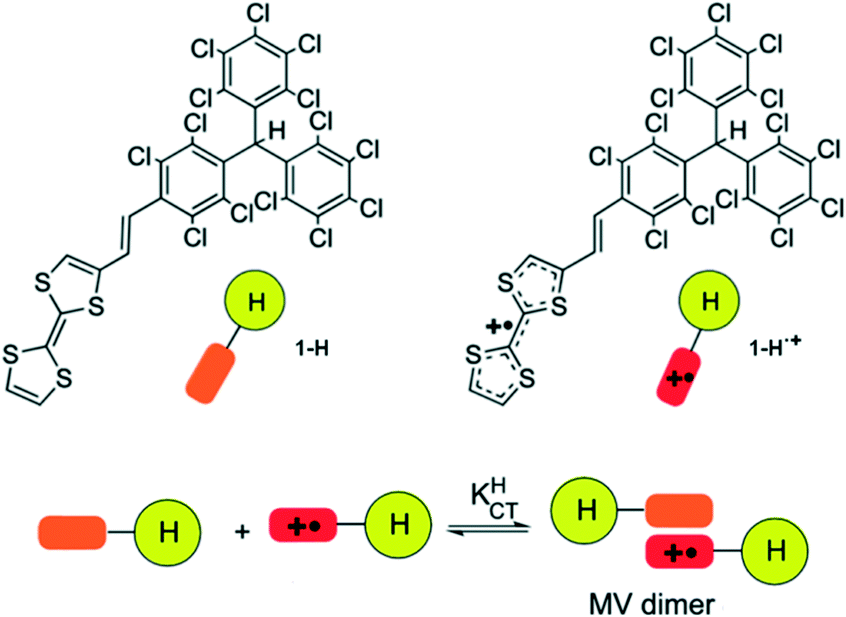 | ||
| Fig. 11 Molecular structures of TTF–PTM dyads with a hydrogenated PTM residue and with the partial oxidation of the TTF subunit. Schematic representation of the formation of the mixed-valence (MV) dimer. | ||
Moreover, a theoretical model, merging a Hubbard-like description of the intermolecular CT interaction with a description of the IET in terms of a minimal model that accounts for just two essential electronic states (neutral and zwitterionic), was proposed in order to rationalize the subtle interplay between the IET within a dyad and the intermolecular CT occurring in a dimer between the TTF residues (Fig. 12).
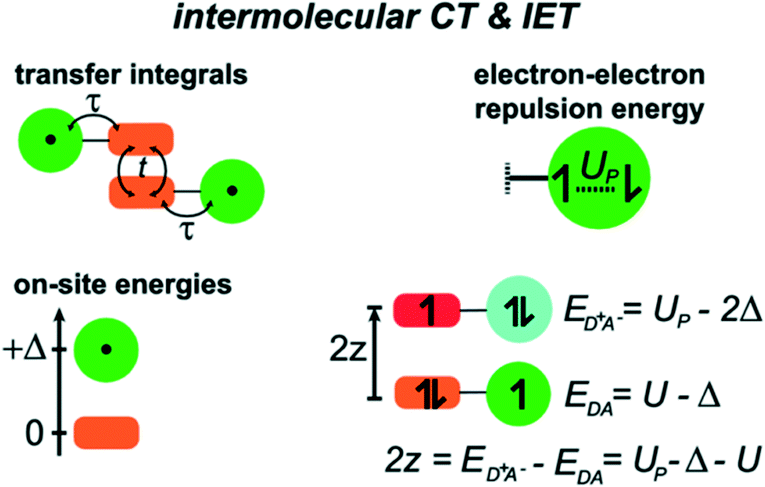 | ||
| Fig. 12 Schematic of the model for intermolecular CT and IET. Adapted with permission from ref. 55. Copyright (2013) American Chemical Society. | ||
Thermomagnetic switch based on a TTF–PTM radical dyad (1)
In order to gain more insight into the dimerization of dyad 1 in DMF, we studied the reversible temperature-induced switching that radical dyad 1 exhibits in DMF solution between diamagnetic dimers [1]2 LT, observed at room temperature, and paramagnetic monomers 1HT at high temperature (Fig. 13).56 This temperature-induced process was followed by different spectroscopic techniques such as UV-vis-NIR, ESR, Raman, proton nuclear magnetic resonance (1H-NMR) and cyclic voltammetry.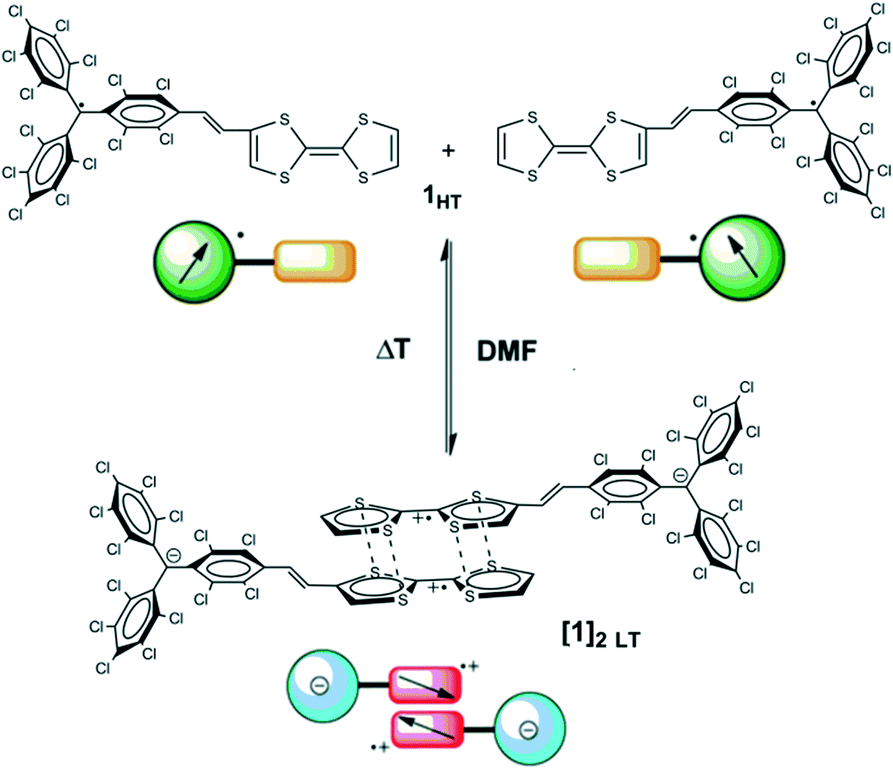 | ||
| Fig. 13 Schematic representation of the reversible temperature-induced supramolecular switching of TTF–PTM (1) between diamagnetic dimers at low temperature ([1]2 LT) and paramagnetic monomers at high temperature (1HT). Adapted with permission from ref. 56. Copyright (2013) American Chemical Society. | ||
Regarding the optical properties, UV-vis-NIR spectra of dyad 1 in DMF were recorded at different temperatures in the 300–375 K range (Fig. 14) observing a clear color change. At 300 K, the spectrum exhibited an intense band at 512 nm assigned to the PTM subunit in its anionic form, indicating that 1 is only present in the zwitterionic D+A− state. When the solution was heated, the band associated with the PTM anion weakens, whereas the band related to the PTM neutral radical (387 nm) acquires intensity together with the appearance of a weak band at 950 nm that is associated with the IET process of 1 in the neutral D–A form.
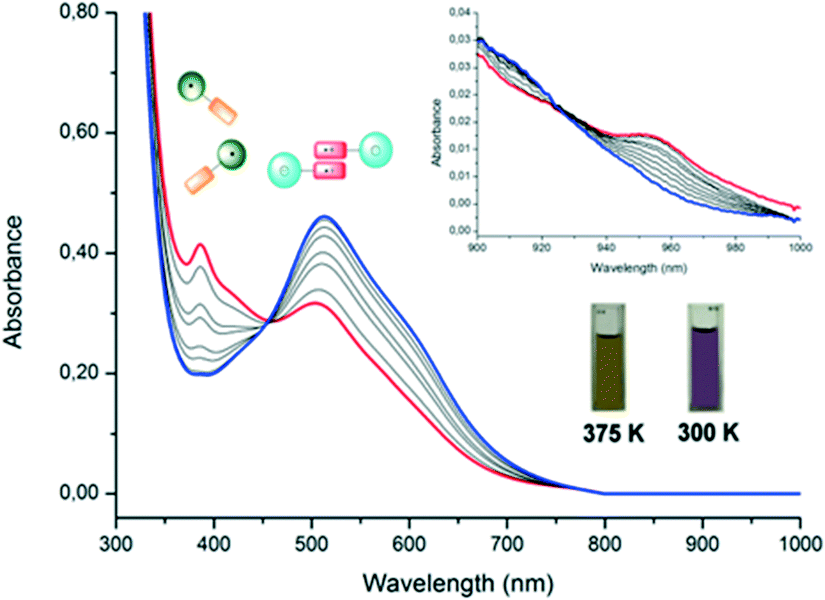 | ||
| Fig. 14 UV-vis spectra of dyad 1 in DMF (0.1 mM) at 375 (red line), 365, 355, 350, 345, 340, 330, 320 and 300 K (blue line). Adapted with permission from ref. 56. Copyright (2013) American Chemical Society. | ||
On the other hand, ESR measurements were performed at different temperatures (Fig. 15). The strong antiferromagnetic coupling of TTF-radical spins in the [1]2 LT dimers is responsible for the lack of ESR signal at room temperature, whereas upon heating the solution at 375 K, a broad line appeared at a g-value of 2.0025. This signal is typical of PTM radicals, indicating the formation of a paramagnetic species that we identified as the 1HT monomer in the neutral D–A form. After cooling down the solution to room temperature, it gradually became ESR silent again, exhibiting a completely reversible behavior from 375 K to 300 K. Moreover, to demonstrate the complete reversibility of the system, several and consecutive ESR and UV-vis cycles at 300 and 375 K were performed without showing any sign of deterioration, highlighting the reversibility to switch the molecular self-assembly in this system.
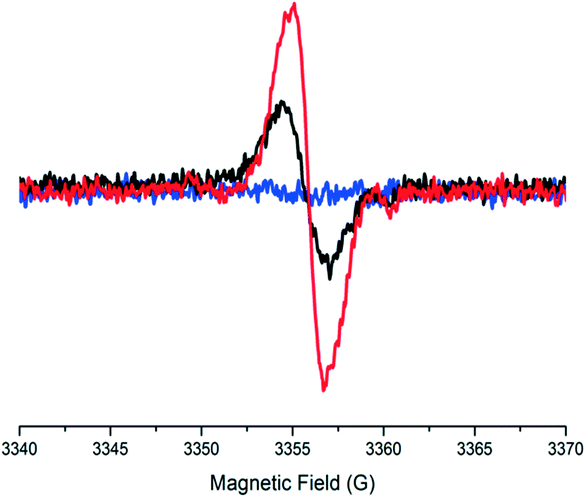 | ||
| Fig. 15 ESR spectra of 1 in DMF (0.1 mM) at 375 (red line), 365 (black line) and 300 (blue line) K. Adapted with permission from ref. 56. Copyright (2013) American Chemical Society. | ||
In view of the interesting electronic properties of some organic acceptor–donor–acceptor (A–D–A) triads,57–59 we have also studied the switching properties of an A–D–A diradical triad based on two PTM radicals connected through a TTF-vinylene bridge (2).60 This molecule exhibited electrochemical reversible switching by a one-electron reduction and oxidation, modifying its optical, magnetic and electronic properties (Fig. 16). Indeed, we studied the generation of the mixed-valence radical anion 2˙− and the triradical cation 2˙˙˙+ species obtained upon electrochemical reduction or oxidation, respectively, monitoring the properties by optical and ESR spectroscopy. Interestingly, modification of the electron delocalization and magnetic coupling was observed when the charged species were generated and these changes were rationalized by theoretical calculations. However, the dimerization process between the TTF moieties taking place in the TTF–PTM dyad was not observed in this case, probably due to the steric hindrance produced by the additional PTM radical moiety.
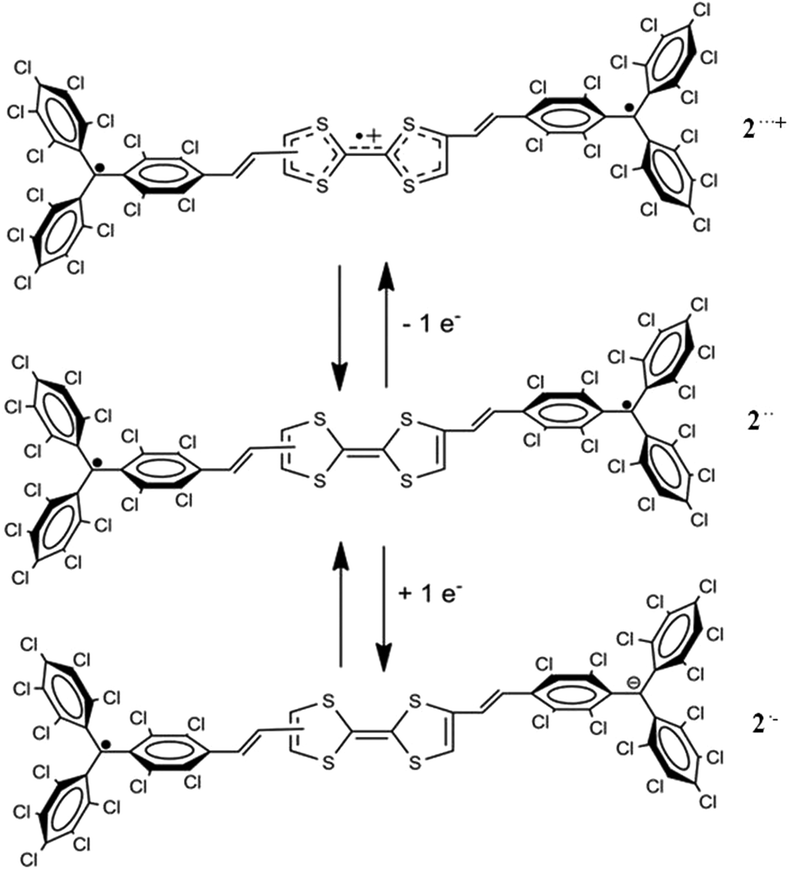 | ||
| Fig. 16 Molecular structures of diradical 2, mixed-valence 2˙− and triradical cation 2˙˙˙+. Adapted with permission from ref. 60. Copyright (2016) American Chemical Society. | ||
Studies in solid state
After the understanding of such interesting physical properties and self-assembling abilities of TTF–PTM dyads in solution, we aimed to extend and exploit these properties in solid state. In fact, one of the main interests of these systems is to combine both the potential conducting properties of the partially oxidized TTF moiety and the magnetic properties of the PTM unit in a cooperative manner in order to develop new spin-polarized donor systems exhibiting magnetoresistance (the change in electrical resistance when applying a magnetic field).61,62 Moreover, the TTF unit can be considered as an ideal building block for improving the intermolecular interactions in order to exploit the conducting properties in the solid state. For this reason, first of all it is necessary to study in detail the crystal structures of different TTF–PTM dyads and to analyze the different supramolecular arrangements.Crystal structure of 1-H
The crystal structure of the hydrogenated derivative 1-H (Fig. 17) showed that it crystallizes in the triclinic system with a P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The molecular structure revealed that the vinylene bridge is in the trans-position and the crystal packing showed that molecules are arranged in a head-to-tail manner with short S⋯Cl (3.38 Å) and Cl⋯Cl (3.35 Å) contacts (Fig. 18). Thus, this supramolecular arrangement was not optimal for developing conducting molecular materials. Unfortunately, to date, it has not been possible to study in detail the properties in the solid state of radical dyad 1 because of the poor crystalline quality which prevents the determination of its X-ray crystal structure.
space group. The molecular structure revealed that the vinylene bridge is in the trans-position and the crystal packing showed that molecules are arranged in a head-to-tail manner with short S⋯Cl (3.38 Å) and Cl⋯Cl (3.35 Å) contacts (Fig. 18). Thus, this supramolecular arrangement was not optimal for developing conducting molecular materials. Unfortunately, to date, it has not been possible to study in detail the properties in the solid state of radical dyad 1 because of the poor crystalline quality which prevents the determination of its X-ray crystal structure.
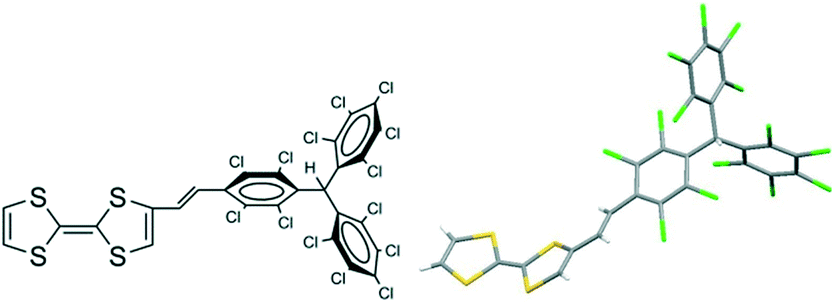 | ||
| Fig. 17 Molecular structure and ORTEP views of 1-H. Atoms are shown at the 50% probability level. | ||
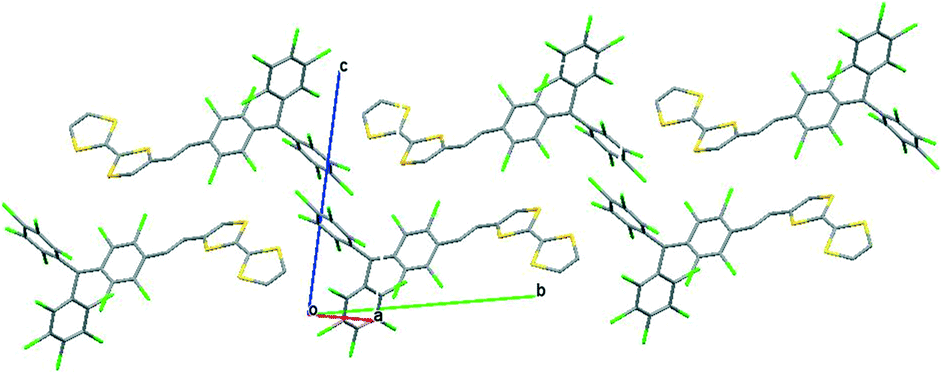 | ||
| Fig. 18 Crystal packing of dyad 1-H in the bc plane. | ||
Crystal structure of the monopyrrolo-TTF (MPTTF)–PTM dyad (3 and 3-H)
In order to improve the self-assembly of the TTF moiety of the TTF–PTM dyads and improve their crystalline quality, we have designed and obtained the TTF-based radical dyad 3 with a longer bridge and higher degree of coplanarity between the TTF and PTM units than dyad 1. The increased length of the bridge was obtained using a MPTTF building block connected through a conjugated phenyl-vinylene to the PTM unit obtaining the MPTTF–PTM dyad 3 and its hydrogenated derivative 3-H (Fig. 19).63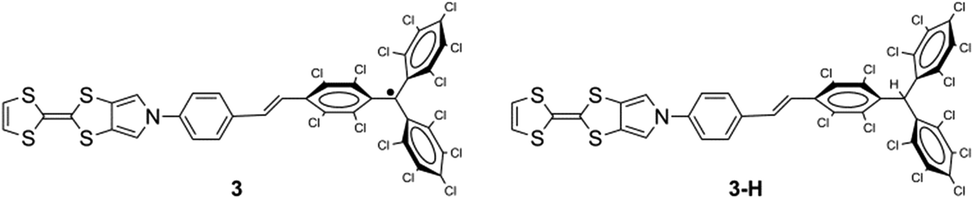 | ||
| Fig. 19 Molecular structures of radical 3 and non-radical 3-H dyads. | ||
X-ray diffraction analysis at 300 K on red crystals of 3-H indicates that it crystallizes in the triclinic system with a P space group. The TTF moieties of neighboring molecules form alternate face-to-face dimers on the bc plane, forming 1-D chains along the c-axis with a shortest S–S distances of 3.92 Å (Fig. 20). On the other hand, PTM units are also stacked forming monodimensional chains along the c-axis with short intermolecular Cl⋯Cl interactions that are in the range of 3.28–3.47 Å. This supramolecular arrangement was not optimal for obtaining conducting materials since the TTF units do not form regular stacks and they show face-to-face dimers with a large space between them.
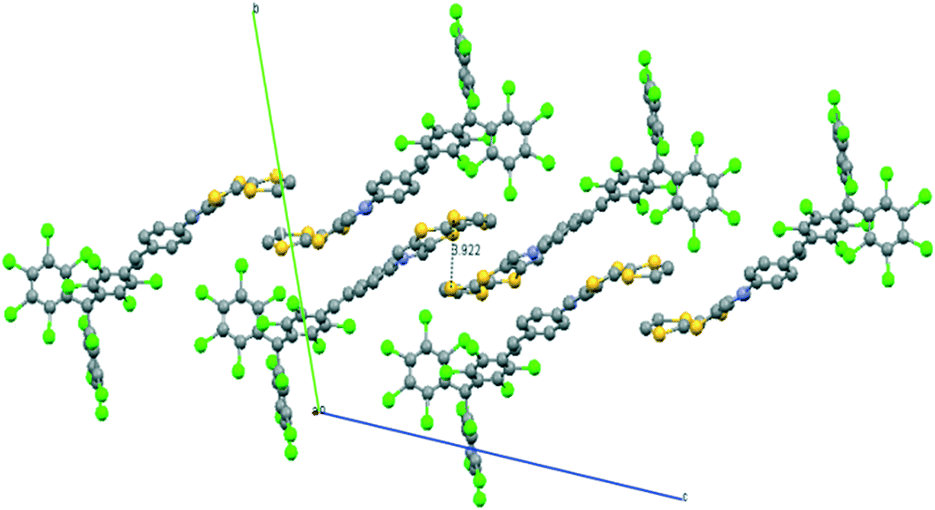 | ||
| Fig. 20 Molecular packing of dyad 3-H in the bc plane. Adapted with permission from ref. 63; Copyright 2015, John Wiley and sons. | ||
On the other hand, the radical dyad 3 crystallizes in the monoclinic system with the space group P21 and the asymmetric unit reveals two inequivalent molecules that are very similar in geometry. Regarding the molecular arrangement, molecules of radical dyad 3 are stacked, forming 1D chains on the ab plane, in which MPTTF units form a herringbone structure with S⋯S and Cl⋯Cl interactions of 3.91 and 3.33 Å, respectively (Fig. 21). In view of such a crystal structure with segregated donor and acceptor units, we proposed that this system could be an optimal candidate for developing a single-component conductor by improving the intermolecular interactions and doping the TTF moieties.
 | ||
| Fig. 21 Molecular packing of radical dyad 3 in the bc plane. Adapted with permission from ref. 63; Copyright 2015, John Wiley and sons. | ||
Molecular radical conductor in solid state
In view of the supramolecular architecture of radical dyad 3 with segregated donor and acceptor units, we aimed to promote conductivity upon the application of high pressure since we could enhance the electronic bandwidth W of the system. Indeed, we have recently reported the resistivity measurements of dyad 3 at high pressure, indicating semiconductor behaviour and a detailed study on the origin of the conductivity at high pressure performed by theoretical calculations as well as Raman spectroscopy at different pressures.64Resistivity measurements of crystals of the radical dyad 3 were performed up to 21 GPa along the b-axis. Crystals of 3 showed insulating behaviour at ambient pressure, whereas they exhibited semiconducting behaviour when increasing the pressure from 6.5 GPa (Fig. 22). The room temperature conductivity of 3 at 15 GPa was found to be 0.76 S cm−1 with a low activation energy (Ea) of 0.067 eV.
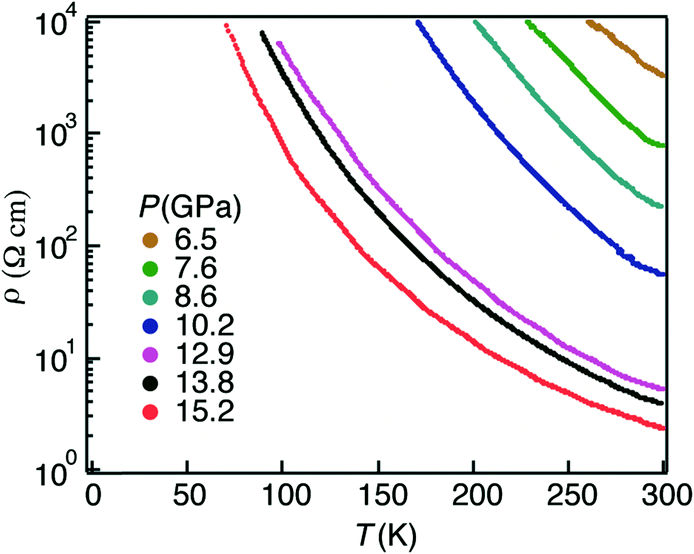 | ||
| Fig. 22 Temperature dependence of the resistivity of radical dyad 3 along the b-axis at different pressures. Adapted with permission from ref. 64. Copyright (2016) American Chemical Society. | ||
On the other hand, crystals of the PTM radical without any substituent exhibited insulating behaviour at all measured pressures. This difference in behaviour was related to the shorter distances between the molecules in the crystal structure of 3 in comparison with unsubstituted PTM due to the short S⋯S intermolecular interactions between the TTF moieties. Thus, the electronic bandwidth W was increased in compound 3 in comparison with the PTM radical without any substituent.
In order to understand the origin of the conducting behaviour in such neutral TTF–PTM dyads, we performed theoretical calculations of the electronic structure and CT based on predicted crystal structures of 3 at high pressure. The spin-polarized band structure calculations showed a gap close to the Fermi level (EF) at ambient pressure between the occupied bands with dominant TTF character (black lines in density of states) and the empty bands with dominant PTM character (Fig. 23). The colours blue and red indicate the different spin orientation. The gap between the bands diminished and closed at pressures higher than 6 GPa; we observed that with increasing pressure the electronic bandwidth W increased by around one order of magnitude (from 0.1 eV at P = 0 GPa to 0.6 eV at P = 8.6 GPa).
 | ||
| Fig. 23 Spin polarized electronic band structures and density of states (DOS) of the radical dyad 3 at 0 GPa and 8.6 GPa. Adapted with permission from ref. 64. Copyright (2016) American Chemical Society. | ||
On the other hand, the pressure evolution of CT was calculated for the different units of the molecule distinguishing the MPTTF moiety as the electron donor and the PTM together with the bridge as the acceptor (Fig. 24). Increasing the pressure, we observed that the CT between the two units increases from 0.09 e− at 0 GPa to 0.32 e− at 18 GPa which is indicative of the partial self-doping ability of the system. This charge reorganization occurring at high pressure could be related to a decrease in the coulombic repulsion energy U of the system when increasing the pressure. Moreover, high-pressure Raman spectroscopy also supported this charge reorganization in a similar pressure regime that was in full agreement with the simulated Raman spectra from the predicted crystal structures at high pressure.
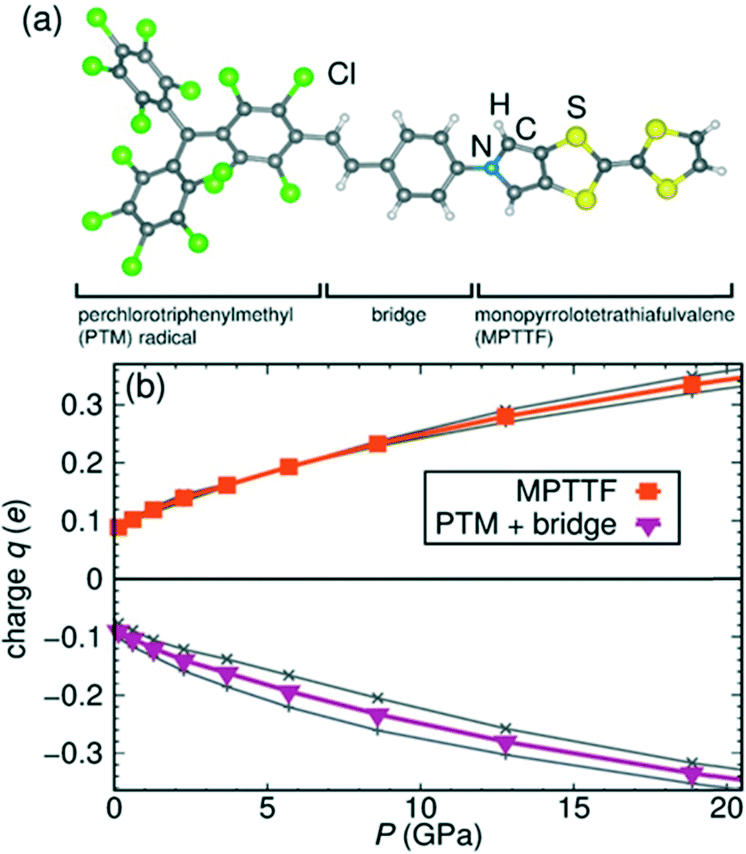 | ||
| Fig. 24 Calculated evolution of excess charge within the units of radical dyad 3 with hydrostatic pressure. (a) The different selected parts of the radical dyad 3. (b) Excess of charge on the different units under high pressure. The grey lines denote the excess charge corresponding to the two non-equivalent molecules of 3 under pressure. Adapted with permission from ref. 64. Copyright (2016) American Chemical Society. | ||
Thus, we have reported the first example of a single-component conductor based on a TTF–π–PTM dyad and using a non-planar spin-localized radical. The origin of the conductivity was mainly attributed to the enhanced intermolecular interactions between the molecules due to the incorporation of the TTF moiety and to the enhanced charge delocalization of the system which acts as internal partial doping. These results have been demonstrated to be a new proof of concept in order to design new neutral radical conductors and it may be possible to engineer new radical dyads which could conduct without the need for applied pressure.
Conclusions and outlook
In summary, we have reported different examples of organic D–π–A dyads based on the electron-donor TTF linked to the electron-acceptor PTM radical, as building blocks, through different conjugated bridges. These TTF–π–PTM dyads showed very interesting physical properties in solution and have been proposed as molecular switches. The interplay between the intra- and intermolecular CT processes plays an important role in the self-assembly of such dyads and the understanding of such a phenomenon is of high interest in order to design molecular conductors in solid state.In the first example, the TTF–PTM radical dyad 1 showed an IET process when tuning the polarity of the solvent. Indeed, the highly polar solvent DMF stabilized the formation of the zwitterionic species and at the same time induced the self-assembly of the molecules forming diamagnetic dimers of zwitterions. Moreover, using temperature as an external stimulus, it was possible to switch the same system between diamagnetic dimers at room temperature and paramagnetic monomers at high temperature that exhibited different optical and magnetic properties.
On the other hand, we extended the study of the self-assembled properties of TTF–π–PTM to the solid state in order to develop new single-component molecular conductors. To this end, we investigated the supramolecular arrangement of different compounds paying a lot of attention to the MPTTF–PTM radical dyad 3 that showed crystal packing with segregated donor and acceptor units with S⋯S interactions between the TTF moieties. In order to promote the conductivity of this system, we applied high pressure to observe the semiconducting behaviour from 6.5 GPa. The origin of such conductivity was attributed to the enhanced electronic bandwidth W due to the short interaction between the molecules and to the charge reorganization occurring in the system, decreasing the Coulombic repulsion U.
These studies demonstrated that organic TTF–π–PTM systems are very interesting materials in the field of molecular electronics, finding applications as molecular switches in solution or conductors in solid state. Tuning the bridge nature and the interplay between the intra- and intermolecular CT offers excellent opportunities for developing new multifunctional molecular materials. Exploring new TTF–π–PTM derivatives could pave the way to exploiting and combining both the optical and magnetic properties in a cooperative manner for applications in spintronics and molecular electronics.
Acknowledgements
This work was supported by the EU Training Networks (ITN) “iSwitch” (GA No. 642196) and “NANO2FUN” (GA No. 607721) DGI (Spain) grant (BeWell; CTQ2013-40480-R), the Networking Research Center on Bioengineering, Biomaterials, and Nanomedicine (CIBER-BBN), and the Generalitat de Catalunya (grant 2014-SGR-17). ICMAB acknowledges support from the Spanish Ministry of Economy and Competitiveness, through the “Severo Ochoa” Programme for Centres of Excellence in R&D (SEV-2015-0496). M. S. is grateful to Spanish Ministerio de Educación, Cultura y Deporte for a FPU grant and he is enrolled in the Material Science Ph.D. program of UAB.Notes and references
- E. Coronado, A. Nuez and F. M. Romero, Solid State Sci., 2003, 5, 917–924 CrossRef CAS
.
-
L. Ouahab, Multifunctional Molecular Materials, CRC Press, 2013 Search PubMed
- G. Poneti, M. Mannini, L. Sorace, P. Sainctavit, M. A. Arrio, E. Otero, J. C. Cezar and A. Dei, Angew. Chem., Int. Ed., 2010, 49, 1954–1957 CrossRef CAS PubMed
- Y. Wang, X. Tan, Y.-M. Zhang, S. Zhu, I. Zhang, B. Yu, K. Wang, B. Yang, M. Li, B. Zou and S. X.-A. Zhang, J. Am. Chem. Soc., 2015, 137, 931–939 CrossRef CAS PubMed
- D. Bléger and S. Hecht, Angew. Chem., Int. Ed., 2015, 2–14 Search PubMed
- A. Droghetti and S. Sanvito, Phys. Rev. Lett., 2011, 107, 2–5 CrossRef PubMed
-
W. R. Feringa and L. Browne, Molecular Switches, Wiley, 2011 Search PubMed
- J. L. Zhang, J. Q. Zhong, J. D. Lin, W. P. Hu, K. Wu, G. Q. Xu, A. T. S. Wee and W. Chen, Chem. Soc. Rev., 2015, 44, 2998–3022 RSC
- A. Bousseksou, G. Molnár, L. Salmon and W. Nicolazzi, Chem. Soc. Rev., 2011, 40, 3313–3335 RSC
- O. Sato, J. Tao and Y.-Z. Z. Zhang, Angew. Chem., Int. Ed., 2007, 46, 2152–2187 CrossRef CAS PubMed
- C. W. Chu, J. Ouyang, J. H. Tseng and Y. Yang, Adv. Mater., 2005, 17, 1440–1443 CrossRef CAS
- D. Small, V. Zaitsev, Y. Jung, S. V. Rosokha, M. Head-Gordon and J. K. Kochi, J. Am. Chem. Soc., 2004, 126, 13850–13858 CrossRef CAS PubMed
- Y. Morita, S. Suzuki, K. Fukui, S. Nakazawa, H. Kitagawa, H. Kishida, H. Okamoto, A. Naito, A. Sekine, Y. Ohashi, M. Shiro, K. Sasaki, D. Shiomi, K. Sato, T. Takui and K. Nakasuji, Nat. Mater., 2008, 7, 48–51 CrossRef CAS PubMed
- Y. Morita, S. Suzuki, K. Sato and T. Takui, Nat. Chem., 2011, 3, 197–204 CrossRef CAS PubMed
- R. G. Hicks, Nat. Chem., 2011, 3, 189–191 CrossRef CAS PubMed
- M. E. Itkis, X. Chi, A. W. Cordes and R. C. Haddon, Science, 2002, 296, 1443–1445 CrossRef CAS PubMed
- S. K. Pal, P. Bag, A. Sarkar, X. Chi, M. E. Itkis, F. S. Tham, B. Donnadieu and R. C. Haddon, J. Am. Chem. Soc., 2010, 132, 17258–17264 CrossRef CAS PubMed
- J. M. Rawson and F. Palacio, Structure, 2001, 100, 94–108 Search PubMed
- J. L. Brusso, O. P. Clements, R. C. Haddon, M. E. Itkis, A. A. Leitch, R. T. Oakley, R. W. Reed and J. F. Richardson, J. Am. Chem. Soc., 2004, 8256–8265 CrossRef CAS PubMed
- R. C. Haddon, Nature, 1975, 256, 394–396 CrossRef CAS
- S. K. Pal, M. E. Itkis, F. S. Tham, R. W. Reed, R. T. Oakley and R. C. Haddon, Science, 2005, 309, 281–284 CrossRef CAS PubMed
- S. K. Mandal, S. Samanta, M. E. Itkis, D. W. Jensen, R. W. Reed, R. T. Oakley, F. S. Tham, B. Donnadieu and R. C. Haddon, J. Am. Chem. Soc., 2006, 128, 1982–1994 CrossRef CAS PubMed
- S. K. Pal, M. E. Itkis, F. S. Tham, R. W. Reed, R. T. Oakley and R. C. Haddon, J. Am. Chem. Soc., 2008, 130, 3942–3951 CrossRef CAS PubMed
- A. Mailman, S. M. Winter, X. Yu, C. M. Robertson, W. Yong, J. S. Tse, R. A. Secco, Z. Liu, P. A. Dube, J. A. K. Howard and R. T. Oakley, J. Am. Chem. Soc., 2012, 134, 9886–9889 CrossRef CAS PubMed
- X. Yu, A. Mailman, K. Lekin, A. Assoud, C. M. Robertson, B. C. Noll, C. F. Campana, J. A. K. Howard, P. A. Dube and R. T. Oakley, J. Am. Chem. Soc., 2012, 134, 2264–2275 CrossRef CAS PubMed
- J. W. L. Wong, A. Mailman, K. Lekin, S. M. Winter, W. Yong, J. Zhao, S. V. Garimella, J. S. Tse, R. A. Secco, S. Desgreniers, Y. Ohishi, F. Borondics and R. T. Oakley, J. Am. Chem. Soc., 2014, 136, 1070–1081 CrossRef CAS PubMed
- L. Yao, S. Zhang, R. Wang, W. Li, F. Shen and B. Yang, Angew. Chem., Int. Ed., 2014, 53, 2119–2123 CrossRef CAS PubMed
- F. Bures, RSC Adv., 2014, 4, 58826–58851 RSC
- R. M. Metzger, Chem. Rev., 2003, 103, 3803–3834 CrossRef CAS PubMed
- A. A. Gorshelev, A. V. Naumov, Y. Eremchev, Y. G. Vainer, L. Kador and J. Kohler, ChemPhysChem, 2010, 11, 182–187 CrossRef CAS PubMed
- I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303 RSC
- Y. Yamashita and M. Tomura, J. Mater. Chem., 1998, 8, 1933–1944 RSC
- J. J. Bergkamp, S. Decurtins and S.-X. Liu, Chem. Soc. Rev., 2015, 44, 863–874 RSC
- Y. Geng, R. Pfattner, A. Campos, J. Hauser, V. Laukhin, J. Puigdollers, J. Veciana, M. Mas-Torrent, C. Rovira, S. Decurtins and S.-X. Liu, Chem. – Eur. J., 2014, 20, 7136–7143 CrossRef CAS PubMed
- J. L. Segura and N. Martn, Angew. Chem., Int. Ed., 2001, 40, 1372–1409 CrossRef CAS
- N. Martín, Chem. Commun., 2013, 49, 7025–7027 RSC
- P. Batail, Chem. Rev., 2004, 104, 4887–4890 CrossRef CAS
- F. Wudl, G. M. Smith and E. J. Hufnagel, J. Chem. Soc. D, 1970, 1453–1454 RSC
- D. Canevet, M. Sallé, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 2245–2269 RSC
- F. Pop and N. Avarvari, Chem. Commun., 2016, 52, 7906–7927 RSC
- G. Ho, J. R. Heath, M. Kondratenko, D. F. Perepichka, K. Arseneault, M. Pézolet and M. R. Bryce, Chem. – Eur. J., 2005, 11, 2914–2922 CrossRef CAS PubMed
- S. V. Rosokha and J. K. Kochi, J. Am. Chem. Soc., 2007, 129, 828–838 CrossRef CAS PubMed
- P. T. Chiang, N. C. Chen, C. C. Lai and S. H. Chiu, Chem. – Eur. J., 2008, 14, 6546–6552 CrossRef CAS PubMed
- M. Yoshizawa, K. Kumazawa and M. Fujita, J. Am. Chem. Soc., 2005, 127, 13456–13457 CrossRef CAS PubMed
-
J. Veciana and I. Ratera, Radicals : Towards Multifunctional Molecular Materials, Wiley, 2010 Search PubMed
- I. Ratera, D. Ruiz-Molina, F. Renz, J. Ensling, K. Wurst, C. Rovira, P. Gütlich and J. Veciana, J. Am. Chem. Soc., 2003, 125, 1462–1463 CrossRef CAS PubMed
- I. Ratera, C. Sporer, D. Ruiz-Molina, N. Ventosa, J. Baggerman, A. M. Brouwer, C. Rovira and J. Veciana, J. Am. Chem. Soc., 2007, 129, 6117–6129 CrossRef CAS PubMed
- G. D'Avino, L. Grisanti, J. Guasch, I. Ratera, J. Veciana and A. Painelli, J. Am. Chem. Soc., 2008, 130, 12064–12072 CrossRef PubMed
- L. Grisanti, G. D'Avino, A. Painelli, J. Guasch, I. Ratera and J. Veciana, J. Phys. Chem. B, 2009, 113, 4718–4725 CrossRef CAS PubMed
- J. Guasch, L. Grisanti, S. Jung, D. Morales, G. D'Avino, M. Souto, X. Fontrodona, A. Painelli, F. Renz, I. Ratera and J. Veciana, Chem. Mater., 2013, 25, 808–814 CrossRef CAS
- M. Souto, D. C. Morales, J. Guasch, I. Ratera, C. Rovira, A. Painelli and J. Veciana, J. Phys. Org. Chem., 2014, 27, 465–469 CrossRef CAS
- G. D'Avino, L. Grisanti, A. Painelli, J. Guasch, I. Ratera and J. Veciana, CrystEngComm, 2009, 11, 2040–2047 RSC
- S. Nishida, Y. Morita, K. Fukui, K. Sato, D. Shiomi, T. Takui and K. Nakasuji, Angew. Chem., Int. Ed., 2005, 44, 7277–7280 CrossRef CAS PubMed
- J. Guasch, L. Grisanti, V. Lloveras, J. Vidal-Gancedo, M. Souto, D. C. Morales, M. Vilaseca, C. Sissa, A. Painelli, I. Ratera, C. Rovira and J. Veciana, Angew. Chem., Int. Ed., 2012, 51, 11024–11028 CrossRef CAS PubMed
- J. Guasch, L. Grisanti, M. Souto, V. Lloveras, J. Vidal-Gancedo, I. Ratera, A. Painelli, C. Rovira and J. Veciana, J. Am. Chem. Soc., 2013, 135, 6958–6967 CrossRef CAS PubMed
- M. Souto, J. Guasch, V. Lloveras, P. Mayorga, J. T. López Navarrete, J. Casado, I. Ratera, C. Rovira, A. Painelli and J. Veciana, J. Phys. Chem. Lett., 2013, 4, 2721–2726 CrossRef CAS
- O. Elsner, D. Ruiz-Molina, J. Vidal-Gancedo, C. Rovira and J. Veciana, Chem. Commun., 1999, 579–580 RSC
- F. Otón, V. Lloveras, M. Mas-Torrent, J. Vidal-Gancedo, J. Veciana and C. Rovira, Angew. Chem., Int. Ed., 2011, 50, 10902–10906 CrossRef PubMed
- J. Calbo, J. Aragó, F. Otón, V. Lloveras, M. Mas-Torrent, J. Vidal-Gancedo, J. Veciana, C. Rovira and E. Ortí, Chem. – Eur. J., 2013, 19, 16656–16664 CrossRef CAS PubMed
- M. Souto, V. Lloveras, S. Vela, M. Fumanal, I. Ratera and J. Veciana, J. Phys. Chem. Lett., 2016, 7, 2334–2339 Search PubMed
- T. Sugawara and M. M. Matsushita, J. Mater. Chem., 2009, 19, 1738 RSC
- T. Sugawara, H. Komatsu and K. Suzuki, Chem. Soc. Rev., 2011, 40, 3105–3118 RSC
- M. Souto, M. V. Solano, M. Jensen, D. Bendixen, F. Delchiaro, A. Girlando, A. Painelli, J. O. Jeppesen, C. Rovira, I. Ratera and J. Veciana, Chem. – Eur. J., 2015, 21, 8816–8825 CrossRef CAS PubMed
- M. Souto, H. Cui, M. Peña, V. G. Baonza, H. O. Jeschke, M. Tomic, R. Valentí, D. Blasi, I. Ratera, C. Rovira and J. Veciana, J. Am. Chem. Soc., 2016, 138, 11517–11525 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2017 |